I riassunti , gli appunti i testi contenuti nel nostro sito sono messi a disposizione gratuitamente con finalità illustrative didattiche, scientifiche, a carattere sociale, civile e culturale a tutti i possibili interessati secondo il concetto del fair use e con l' obiettivo del rispetto della direttiva europea 2001/29/CE e dell' art. 70 della legge 633/1941 sul diritto d'autore
Le informazioni di medicina e salute contenute nel sito sono di natura generale ed a scopo puramente divulgativo e per questo motivo non possono sostituire in alcun caso il consiglio di un medico (ovvero un soggetto abilitato legalmente alla professione).
L'esistenza di aggregati di materia allo stato solido e liquido ci induce a ritenere che esistano delle forze anche tra molecole neutre in grado di legarle. Tali forze si producono sia tra molecole polari che tra molecole apolari e sono conosciute come forze di van der Waals. Le forze o interazioni di van der Waals, note anche come interazioni di non legame o legami deboli (definizione ambigua poichè in fisica le interazioni deboli sono una delle quattro forze fondamentali di natura), hanno un’intensità (0.1 - 10 kJ mol–1) che è mediamente di circa due ordini di grandezza inferiore all’intensità di un legame covalente o ionico (100 - 1000 kJ mol–1). Inoltre tali forze hanno un raggio d’azione estremamente breve, indebolendosi rapidamente all’aumentare della distanza.
L’energia di tali legami è infatti inversamente proporzionale alla sesta potenza della distanza che separa le particelle interagenti (E µ 1/r6), mentre le forze di van der Waals decrescono secondo la settima potenza della distanza (FvdWµ 1/r7).
Esistono tre tipi di forze di van der Waals:
Le molecole polari, o dipoli permanenti (molecole dotate di un momento di dipolo m), esercitano naturalmente una reciproca attrazione elettrostatica. Quando le molecole dipolari si avvicinano tendono infatti a disporsi con i poli di carica opposta l'uno di fronte all'altro, al fine di rendere minima l'energia potenziale del sistema (configurazione di maggior stabilità). In tal modo si verifica un'attrazione elettrostatica tra i poli opposti, detta interazione dipolo-dipolo. Le forze di Keesom agiscono dunque tramite un effetto di orientazione.
Le interazioni dipolo-dipolo non sono molto efficienti finché le molecole si trovano allo stato aeriforme poiché le distanze intermolecolari sono troppo elevate. Finché la temperatura è sufficientemente elevata e/o la pressione bassa, l'energia cinetica media dei dipoli è in grado di vincere tali interazioni, mantenendo la sostanza allo stato aeriforme. Ma all'abbassarsi della temperatura e/o all’aumentare della pressione, le distanze intermolecolari diminuiscono e l'energia cinetica media delle molecole finisce per diventare minore delle interazioni dipolari. In queste condizioni tali forze sono in grado di mantenere adese le molecole favorendo il passaggio ad una fase condensata (liquida o solida). Inizialmente si ha il passaggio allo stato liquido e, se la temperatura scende ulteriormente (o la pressione aumenta), le forze di Keesom sono in grado di bloccare le molecole in posizioni di equilibrio all'interno di un reticolato solido.

Interazioni dipolo-dipolo tra molecole polari allo stato solido
Le interazioni dipolo-dipolo sono ovviamente tanto più intense quanto maggiore è il momento di dipolo m ed iniziano a diventare importanti per valori di m superiori ad 1 D. La loro intensità decresce all’aumentare della temperatura, poiché una maggior agitazione termica interferisce con l’allineamento dei dipoli.
Per due dipoli liberi di ruotare (liquido o aeriforme) di momento m1 ed m2 a distanza r l’energia di Keesom è
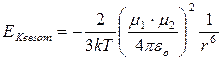
con
k = costante di Boltzmann = 1,38 10-23 J K-1
εo = costante delettrica del vuoto = 8,854 10-12 C2 m-2 N-1
T = temperatura assoluta
Ad esempio, a 25°C l’energia di interazione per una coppia di molecole con μ = 1 D (= 3,336 10-30 C m) alla distanza di 0.3 nm (= 3 10-10 m) è di
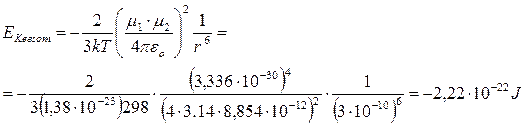
L’energia di Keesom per una mole si ottiene moltiplicando il risultato precedente per il numero di Avogadro N = 6.022 1023 mol-1.
- 2,22 10-22 x 6.022 1023 = - 1340 J mol-1 = - 1,34 kJ mol-1
Per due dipoli stazionari (all’interno di un solido) l’energia di Keesom risulta inversamente proporzionale alla terza potenza della distanza r e dipende dall’orientazione reciproca (angoli θ e φ).
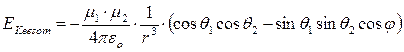
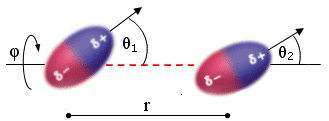
Quando il dipolo è costituito da un atomo di idrogeno legato con legame covalente fortemente polare ad un elemento molto elettronegativo (F, O, N), il legame dipolo-dipolo è particolarmente intenso e viene chiamato legame a idrogeno (o ponte idrogeno). I legami a idrogeno presentano energie tipiche superiori (20 – 50 kJmol-1) rispetto ai normali legami dipolo-dipolo.
Il legame a idrogeno viene rappresentato con una breve linea tratteggiata che unisce l'idrogeno di una molecola con l'elemento elettronegativo di un'altra.
Tipici composti in grado di dare intensi legami a idrogeno sono l'acido fluoridrico HF, l'acqua H2O e l'ammoniaca NH3.
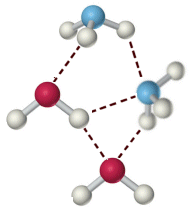
Legami a idrogeno tra molecole d’acqua e ammoniaca
L'esistenza di tale legame aumenta notevolmente la coesione interna tra le molecole, al punto da riflettersi in modo evidente su alcune proprietà fisiche delle sostanze interessate.
Ad esempio tutti i composti le cui molecole sono interessate dai legami a idrogeno presentano temperature di ebollizione e capacità termiche particolarmente elevate.
Se infatti forniamo calore ad una sostanza produciamo un aumento della sua energia cinetica media (½mv²). E' allora evidente che a parità di calore fornito l'aumento di velocità sarà minore per le molecole più massicce. Poiché inoltre una sostanza è in grado di passare allo stato di vapore quando le sue molecole sono sufficientemente veloci, dobbiamo attenderci che la temperatura di ebollizione di un composto sia tanto maggiore quanto maggiore è il suo peso molecolare.
Tale previsione è verificabile osservando ad esempio i composti dell'idrogeno con gli elementi del VII gruppo A, dove il punto di ebollizione diminuisce costantemente al diminuire del peso molecolare, con la notevole eccezione dell'acido fluoridrico.
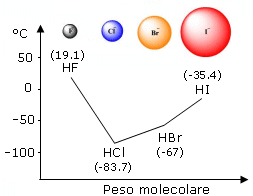
In questo caso infatti, nonostante il basso peso molecolare, la temperatura di ebollizione risulta particolarmente elevata in quanto per poter passare allo stato di vapore le molecole devono possedere un'energia cinetica molto elevata per rompere i legami a idrogeno che le tengono adese.
La presenza del legame a idrogeno spiega anche perchè il ghiaccio sia meno denso dell'acqua. Infatti quando l'acqua si solidifica i legami a idrogeno tendono a bloccare le molecole in una struttura esagonale ordinata che risulta meno densa della struttura disordinata caratteristica dell'acqua liquida.
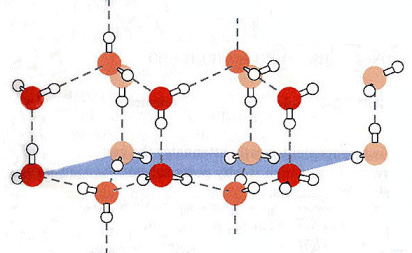
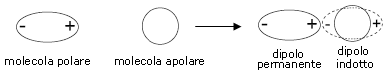
Le forze di Debye si originano tra molecole polari e molecole apolari. Per comprendere tali interazioni è necessario esaminare ciò che accade ad una molecola (o un atomo) apolare quando viene posta in un campo elettrico E.
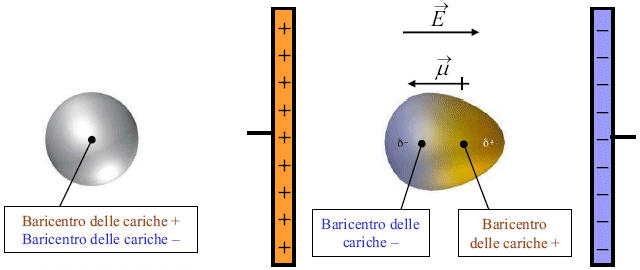
a) molecola non polarizzata b) Polarizzazione in un campo elettrico E
La nuvola elettronica della molecola viene deformata ed attratta dal polo positivo. Il campo elettrico induce dunque una separazione di carica con formazione di un dipolo indotto. L’intensità del momento di dipolo indotto m è direttamente proporzionale all’intensità E del campo elettrico applicato.
m = a E
La costante di proporzionalità a è detta polarizzabilità. Il valore della polarizzabilità è caratteristico per ciascun atomo o molecola ed è una misura della facilità con cui la nuvola elettronica può essere deformata (polarizzata) da un campo elettrico.
L'unità di misura della polarizzabilità nel Sistema Internazionale è C·m2·V-1.
Spesso invece della polarizzabilità si usa il volume di polarizzabilità α', definito da:
![]()
dove εo è la costante dielettrica del vuoto.
α' ha le dimensioni di un volume. L'unità di misura utilizzata nella pratica comune per esprimere il suo valore è il cm3 o Å3.
La polarizzabilità dipende dalla forza con cui gli elettroni esterni sono vincolati al nucleo (minore è l’energia di ionizzazione, maggiore è la polarizzabilità). In altre parole, la nuvola elettronica è tanto più facilmente polarizzabile quanto minore è la forza di attrazione che il nucleo esercita su di essa. La polarizzabilità aumenta all’aumentare della massa e delle dimensioni della molecola.
Quando una molecola polare si avvicina ad una non polare induce in quest'ultima un dipolo eletrico di minore intensità (effetto di induzione) che perdura fintanto che le due molecole restano vicine. Si genera così un’attrazione dipolo permanente-dipolo indotto . L'intensità è proporzionale al momento del dipolo permanente m che induce la polarizzazione e alla polarizzabilità a della seconda molecola.
catione
Per un dipolo di momento m ed una molecola apolare di polarizzazione a a distanza r l’energia di Debye è
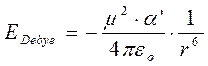
con εo = costante delettrica del vuoto = 8,854 10-12 C2 m-2 N-1
Ad esempio, a 25°C l’energia di interazione per un dipolo (ad esempio HCl) con μ = 1 D (= 3,336 10-30 C m) ed una molecola apolare (ad esempio il benzene) con polarizzazione a’ = 10-23 cm3 (= 10-29 m3) alla distanza di 0.3 nm (= 3 10-10 m) è di
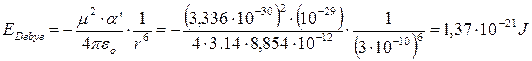
L’energia di Debye per una mole si ottiene moltiplicando il risultato precedente per il numero di Avogadro N= 6.022 1023 mol-1.
- 1,37 10-21 x 6.022 1023 = - 820 J mol-1 = - 0,82 kJ mol-1
Se anche le molecole perfettamente apolari come O2 e Cl2 sono in grado di liquefare e solidificare a temperature superiori allo zero assoluto, evidentemente devono esistere anche per tali sostanze delle forze intermolecolari, seppur molto deboli, in grado di vincere l'agitazione termica.
Si ritiene che tali forze, dette forze di London o forze di dispersione, siano dovute a fluttuazioni temporanee e casuali nella distribuzione di densità degli orbitali. In una molecola apolare la nuvola elettronica è “in media” distribuita in modo omogeneo, ma in un determinato istante questo può non essere vero e gli elettroni possono casualmente e temporaneamente essere addensati a formare un dipolo istantaneo (o dipolo momentaneo o dipolo temporaneo).

Piccole fluttuazioni nella distribuzione delle nuvole elettroniche dovrebbero essere dunque in grado di produrre momentanee polarità anche nelle molecole apolari capaci di indurre nelle molecole adiacenti polarità di segno contrario (dipolo indotto), creando in definitiva le condizioni per un'attrazione reciproca.
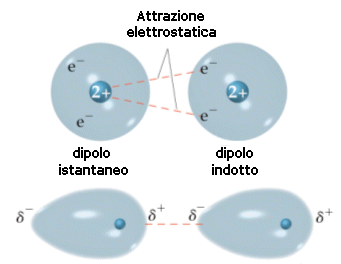
Se si considera la media nel tempo, la nuvola elettronica di un atomo è perfettamente simmetrica, ma in un dato istante può addensarsi maggiormente da un lato ed in un istante immediatamente successivo può spostarsi all’altra estremità. Ciò determina la comparsa di un momento di dipolo elettrico istantaneo variabile nel tempo e mediamente nullo. Ciascun dipolo istantaneo genera un campo elettrico che polarizza le particelle circostanti, creando dei “dipoli indotti” variabili continuamente. Tra il dipolo “induttore” e il dipolo “indotto” nascono così forze di attrazione.
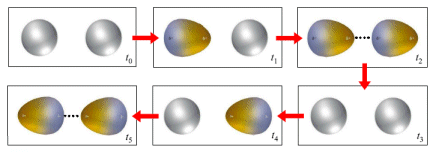
L’intensità delle forze di London dipende ovviamente solo dalla polarizzabilità a delle molecole. Molecole più grandi e massicce (con elettroni superficiali meno legati) risentono in misura maggiore delle forze di London. Ad esempio, a temperatura ambiente, mentre F2 e Cl2 sono gassosi, Br2 è liquido e I2 è solido.

Per due molecole apolari aventi polarizzazione a1 ed a2 a distanza r l’energia di London è
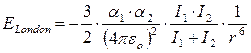
dove
I = hn è l’energia di ionizzazione
con
h = costante di Planck = 6.626 10-34 J s
n= frequenza principale di assorbimento della molecola
Per due molecole identiche la relazione diventa
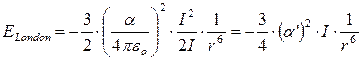
Nel caso di due molecole di metano, ad esempio, con α' = 2.6·10-30 m3 e I = 7 eV (= 1,12·10-18 J) l’energia di dispersione alla distanza di 0.3 nm (= 3·10-10 m) è di
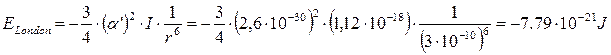
L’energia di London per una mole si ottiene moltiplicando il risultato precedente per il numero di Avogadro N= 6.022 1023 mol-1.
- 7,79·10-21 x 6.022·1023 = - 4690 J mol-1 = - 4,69 kJ mol-1
Le forze di London sono universali, essendo presenti anche in tutti i tipi di atomi e molecole e, nella maggior parte dei casi, costituiscono la componente prevalente delle forze di van der Waals anche in molecole polari. Affinché le interazioni dipolo-dipolo (forze di Keesom) inizino ad essere significative prevalenti rispetto alle forze di London è infatti necessario che le molecole siano di piccole dimensioni (poco polarizzabili) ed abbiano un momento di dipolo superiore ad 1 D (esempi tipici sono l’acqua e l’ammoniaca). Le forze di induzione (forze di Debye) risultano invece per lo più trascurabili.
|
μ (10-30 C m) |
α' (10-30 m3) |
En Ionizz. (eV) |
Keesom (orientazione) kJ mol-1 (%) |
Debye (induzione) kJ mol-1 (%) |
London (dispersione) kJ mol-1 (%) |
Ar |
0 |
1.63 |
15.4 |
0 (0%) |
0 (0%) |
4 (100%) |
CO |
0.40 |
1.99 |
14.3 |
2,4·10-3 (~0%) |
2,4·10-3 (~0%) |
5,62 (99,9%) |
HCl |
3.50 |
2.63 |
13.7 |
1,5 (13%) |
0,23 (2%) |
9,41 (85%) |
HBr |
2.67 |
3.61 |
13.3 |
0,49 (3%) |
0,18 (1%) |
17,2 (96%) |
HI |
1.40 |
5.44 |
12 |
2,8·10-2 (0,1%) |
6,5·10-2 (0,2%) |
35,2 (99,7%) |
NH3 |
4.87 |
2.26 |
16 |
6,6 (44%) |
0,4 (2%) |
8,11 (54%) |
H2O |
6.17 |
1.59 |
18 |
15,7 (76%) |
0,42 (2%) |
4,52 (22%) |
Valori calcolati a 25°C per una distanza intermolecolare di 0,3 nm (3 Å)
|
||||||
In generale, l’interazione attrattiva totale fra molecole neutre, polari o apolari, denominata attrazione di van der Waals, dipende dal contributo delle interazioni descritte precedentemente, dipolare (di orientazione), di induzione e di dispersione. Dato che nella fase fluida tutte tre dipendono dall’inverso della sesta potenza della distanza si può esprimere unitariamente l’energia di attrazione come
![]()
dove la costante di proporzionalità A dipende dalla natura delle molecole interagenti.
Le forze di attrazione di van der Waals prevalgono alle distanze intermolecolari maggiori. Esse hanno un range compreso fra qualche Ǻ ed un centinaio di Ǻ.
A distanze inferiori di qualche Ǻ entrano tuttavia in gioco forze repulsive. L’effetto repulsivo a corto raggio, chiamata repulsione sterica o repulsione di van der Waals, si genera tra i nuclei che, a distanze piccole, non sono più ben schermati dagli elettroni, e fra gli elettroni stessi, soggetti a una forza repulsiva che si genera quando due o più di essi tendono ad occupare gli stessi numeri quantici, in opposizione al principio di Pauli.
Tali forze repulsive, caratterizzate da un raggio d'azione molto breve, crescono rapidamente all'avvicinarsi delle molecole. Il calcolo delle interazioni tra coppie di molecole a brevi distanze presenta notevoli difficoltà. Per esse non esiste infatti un'equazione ricavata teoricamente che le descriva e ci si affida quindi ad alcune funzioni potenziali empiriche. E’ richiesto solo che esse tendano a zero per r che tende all’infinito più velocemente del termine r6.
Una drastica approssimazione, che in qualche caso si adotta per semplificare la trattazione, consiste nell'assumere per le repulsioni a corto raggio la forma
E = 0 per r > s
E = ∞ per r ≤ s
note come repulsioni a sfera rigida (hard-sphere).
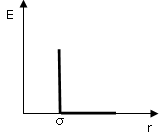
dove σ, detto diametro di collisione, è il diametro della sfera che approssima l'atomo o la molecola. È la distanza di massimo avvicinamento di due molecole e dunque la distanza che separa i due nuclei quando le molecole urtano ed al di sotto della quale la repulsione diventa infinita (impenetrabilità).
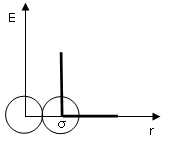
Per determinare s si utilizza spesso la somma dei raggi di van der Waals delle molecole interagenti.
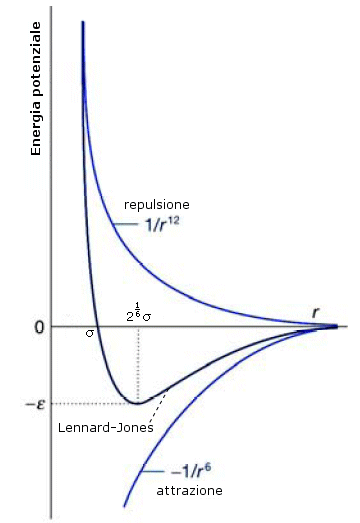
Una approssimazione migliore e molto utilizzata rispetto al modello a sfera rigida è quella di Lennard-Jones in cui la parte repulsiva ha una dipendenza da r12
![]()
dove la costante di proporzionalità B dipende, ancora una volta, dalla natura delle molecole. La repulsione cresce inversamente alla distanza r fra gli atomi elevata alla dodicesima potenza, cioè molto bruscamente. Non esistono argomenti teorici in favore dell’esponente 12, che è stato scelto solo per convenienza di calcolo (essendo il quadrato del termine r6).
Per ottenere l’energia netta dell’interazione intermolecolare si sommano le energie di repulsione e di attrazione viste precedentemente
![]()
Questa relazione si scrive solitamente nella forma del potenziale di Lennard-Jones
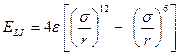
in cui compaiono i parametri σ e ε, il cui valore dipende dal tipo di atomi.
Il primo (σ) ha le dimensioni di una lunghezza ed è la distanza alla quale il potenziale si annulla.
Il secondo (ε) ha le dimensioni di un'energia e rappresenta la profondità della buca di potenziale e dunque l’energia di interazione intermolecolare.
I due parametri possono essere messi in relazione rispettivamente con il diametro atomico e con la massima energia di attrazione tra una coppia di molecole. Se A e B sono molecole diverse, si definisce
![]()
![]() .
.
Il potenziale di Lennard-Jones è particolarmente adatto per simulazioni di gas nobili
Fonte: http://www.pianetachimica.it/didattica/documenti/Chimica_Generale.doc
Sito web da visitare: http://www.pianetachimica.it
Autore del testo: non indicato nel documento di origine
Il testo è di proprietà dei rispettivi autori che ringraziamo per l'opportunità che ci danno di far conoscere gratuitamente i loro testi per finalità illustrative e didattiche. Se siete gli autori del testo e siete interessati a richiedere la rimozione del testo o l'inserimento di altre informazioni inviateci un e-mail dopo le opportune verifiche soddisferemo la vostra richiesta nel più breve tempo possibile.
I riassunti , gli appunti i testi contenuti nel nostro sito sono messi a disposizione gratuitamente con finalità illustrative didattiche, scientifiche, a carattere sociale, civile e culturale a tutti i possibili interessati secondo il concetto del fair use e con l' obiettivo del rispetto della direttiva europea 2001/29/CE e dell' art. 70 della legge 633/1941 sul diritto d'autore
Le informazioni di medicina e salute contenute nel sito sono di natura generale ed a scopo puramente divulgativo e per questo motivo non possono sostituire in alcun caso il consiglio di un medico (ovvero un soggetto abilitato legalmente alla professione).
"Ciò che sappiamo è una goccia, ciò che ignoriamo un oceano!" Isaac Newton. Essendo impossibile tenere a mente l'enorme quantità di informazioni, l'importante è sapere dove ritrovare l'informazione quando questa serve. U. Eco
www.riassuntini.com dove ritrovare l'informazione quando questa serve