I riassunti , gli appunti i testi contenuti nel nostro sito sono messi a disposizione gratuitamente con finalità illustrative didattiche, scientifiche, a carattere sociale, civile e culturale a tutti i possibili interessati secondo il concetto del fair use e con l' obiettivo del rispetto della direttiva europea 2001/29/CE e dell' art. 70 della legge 633/1941 sul diritto d'autore
Le informazioni di medicina e salute contenute nel sito sono di natura generale ed a scopo puramente divulgativo e per questo motivo non possono sostituire in alcun caso il consiglio di un medico (ovvero un soggetto abilitato legalmente alla professione).
Da un punto di vista fisico la risonanza è essenzialmente un modo per descrivere un fenomeno di delocalizzazione che interessa i sistemi coniugati.
Un sistema coniugato è costituito da un orbitale p su di un atomo adiacente ad un legame π (legame doppio o triplo). Vi sono 4 possibili configurazioni per un sistema coniugato:
1) l’orbitale p è coinvolto anch’esso in un legame π (doppi legami coniugati)
– X = Y – Z = W–
2) L’orbitale p è vuoto (tipicamente, ma non necessariamente, l’atomo che lo porta è carico positivamente)
– X = Y – Z +
3) L’orbitale p è saturo (tipicamente, ma non necessariamente, l’atomo che lo porta è carico negativamente)
– X = Y – Z:–
4) L’orbitale p è semisaturo (radicale coniugato)
– X = Y – Z ·
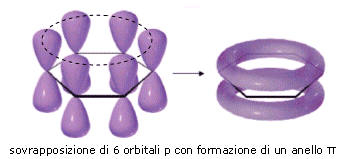
Quando si presenta una di queste configurazioni, vi sono le condizioni affinché si produca un fenomeno di delocalizzazione elettronica (rappresentabile attraverso strutture di risonanza) tra gli atomi del sistema coniugato. I due orbitali p del doppio legame e l’orbitale p dell’atomo adiacente risultano infatti sovrapposti portando ad una delocalizzazione degli elettroni su 3 atomi (4, nel caso di doppi legami coniugati). Tutti gli atomi coinvolti nella delocalizzazione risultano ibridati sp2 (o sp) e quindi planari, con gli orbitali p disposti perpendicolarmente al piano d’ibridazione e parallelamente l’uno rispetto all’altro. La complanarità degli atomi coinvolti, con gli orbitali p disposti parallelamente, è essenziale affinchè gli orbitali p possano sovrapporsi e dar luogo alla delocalizzazione e quindi alla risonanza.
Se ad esempio analizziamo la struttura di Lewis dell’etenammina senza considerare la risonanza saremmo indotti a ritenere che l’atomo di Azoto sia ibridato sp3 (presenta 3 legami ed un doppietto solitario)
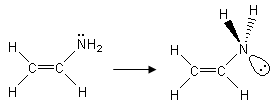
In realtà l’etenammina è una molecola perfettamente planare, come ci suggerisce la sua seconda struttura di risonanza in cui è presente un legame doppio C=N
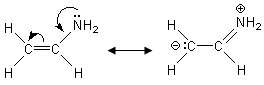
Anche l’Azoto è dunque ibridato sp2 in modo che il suo orbitale p, contenente il doppietto solitario, sia parallelo agli orbitali p dei due atomi di carbonio e si possa sovrapporre ad essi.

La presenza di gruppi chimici ingombranti che impediscano il parallelismo tra gli orbitali p, inibisce il fenomeno della risonanza (inibizione sterica della risonanza).
In alcuni casi tale condizione può essere graficamente rappresentata senza ricorrere alle formule limite. Ad esempio l'anidride solforosa può essere rappresentata anche così
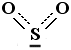 oppure
oppure 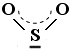
con gli elettroni π delocalizzati su tutta la molecola indicati dalla linea tratteggiata.
A) Sistema coniugato π- π
Nel rappresentazione tradizionale (non delocalizzata) di doppi legami coniugati (–X=Y–Z=W–) 2 doppi legami π si trovano separati da un legame semplice σ. Gli orbitali p si sovrappongono 2 a 2 ed è sufficiente un’unica formula di struttura per descrivere la molecola
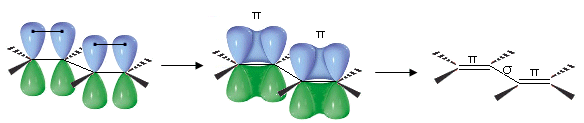
In realtà il sistema coniugato presenta i 4 orbitali p tra loro completamente sovrapposti e gli elettroni π risultano pertanto delocalizzati su 4 atomi.
Per rappresentare la delocalizzazione si utilizzano più formule di struttura, in cui il doppio legame si trova anche in posizione centrale. Il sistema coniugato π-π viene rappresentato con le seguenti tre strutture di risonanza
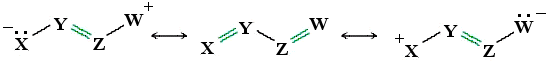
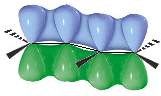
Un importante esempio di delocalizzazione elettronica in un sistema coniugato π-π si ha nel benzene C6H6. un composto organico in cui i 6 atomi di carbonio si chiudono a formare un esagono. Ciascun atomo di carbonio è ibridato sp2 ed impegna i tre orbitali sp2 per legarsi ad un idrogeno e ad altri due atomi di carbonio. L'orbitale pz non ibridato viene utilizzato per formare un legame π con un carbonio adiacente. Si dovrebbe pertanto ritenere che i 6 atomi di carbonio siano uniti all'interno dell'anello da una serie di legami semplici alternati a legami doppi.
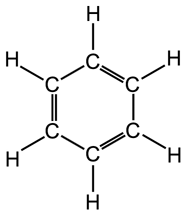
In realtà i sei legami C - C risultano essere perfettamente identici e a metà strada tra un legame semplice ed un legame doppio.
Descriviamo dunque il benzene come un ibrido di risonanza delle due seguenti strutture limite
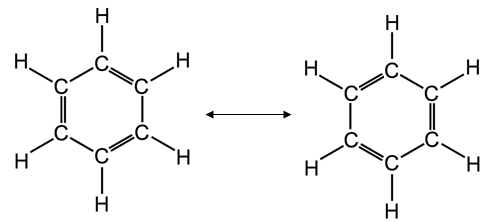
oppure, in modo del tutto equivalente, rappresentiamo le tre coppie di elettroni delocalizzati su tutta la molecola con un anello interno all'esagono
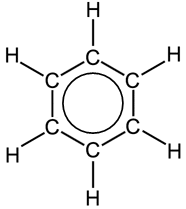
B) Sistema coniugato π-p
In un sistema coniugato π-p (–X=Y–Z*) l’orbitale p adiacente al doppio legame non è impegnato in alcun legame e può contenere da 0 a 2 elettroni (* = 0, 1, 2 elettroni)

Anche in questo caso il sistema coniugato presenta in realtà i 3 orbitali p tra loro completamente sovrapposti e gli elettroni risultano pertanto delocalizzati su 3 atomi.
Il sistema coniugato π-p viene rappresentato con due strutture di risonanza
![]()
A conferma di ciò i due legami non presentano la lunghezza tipica di un legame semplice ed uno doppio, ma hanno una lunghezza intermedia.
Si veda ad esempio la molecola dell’Ozono O3
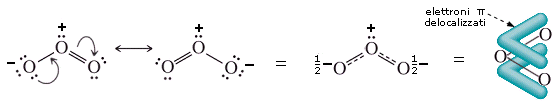
I due legami O-O hanno una lunghezza di 128 nm, intermedia tra quella di un legame semplice O-O (149 nm) e quella di un legame doppio O=O (121 nm).
Nel caso l’orbitale p coniugato al doppio legame contenga una carica positiva o negativa, anche quest’ultima risulta delocalizzata.
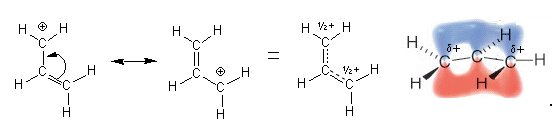
Nel carbocatione allilico, ad esempio, la carica positiva è delocalizzata essendo portata per metà in C1 e per metà in C3
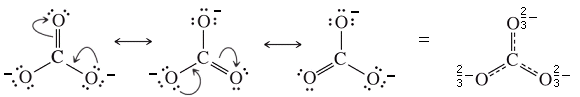
Nell’anione carbonato CO32-le due cariche negative sono distribuite su tutti e tre gli atomi di ossigeno
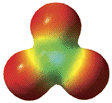
La mappa di potenziale elettrostatico dell’anione carbonato ci conferma la distribuzione di carica.
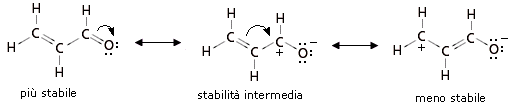
Abbiamo visto che le strutture-limite più stabili contribuiscono maggiormente all’ibrido. In altre parole l’ibrido assomiglia di più alle sue strutture-limite più stabili. Per valutare la stabilità relativa delle diverse strutture di risonanza si applicano, in ordine di importanza, i seguenti 4 criteri
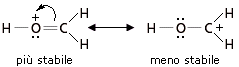
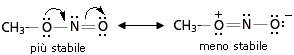
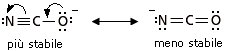
La dimensione degli atomi va generalmente valutata per atomi che appartengano al medesimo gruppo chimico, aventi quindi la medesima configurazione elettronica superficiale, ma diversa dimensione (il raggio atomico aumenta scendendo lungo un gruppo). Le cariche negative sono meglio “sopportate” da atomi più grandi, che riescono in tal modo a disperderle su di un maggior volume atomico diminuendo la densità di carica. Le cariche positive sono meglio “sopportate” da atomi più piccoli, in cui gli elettroni di valenza, trovandosi più vicini al loro nucleo, risultano più saldamente legati.
La struttura più stabile per l’anione S-metantioato è quella di sinistra poiché la carica negativa è portata dallo Zolfo che, pur essendo meno elettronegativo dell’Ossigeno, presenta dimensioni atomiche maggiori.
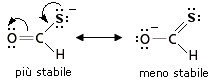
Infine le cariche negative sono meglio sopportate da atomi in cui l’ibridazione ha un maggior carattere s. Un’orbitale sp, avendo un 50 % di carattere s, tiene gli elettroni più vicini al nucleo di un orbitale sp2 (33% di carattere s) o di un orbitale sp3 (25% di carattere s). Nell’esempio seguente la struttura più stabile è quella di destra
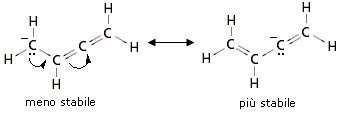
Come abbiamo visto la teoria del Legame di Valenza descrive il legame chimico attraverso le seguenti ipotesi:
La teoria VB si trova tuttavia in difficoltà nello spiegare le proprietà magnetiche di molte molecole semplici (O2) e nel descrivere gli stati eccitati delle molecole e quindi nell’interpretare le proprietà spettroscopiche.
La teoria dell’orbitale molecolare è una teoria quantomeccanica del legame covalente che permette di descrivere lo stato di legame di molecole che la teoria VB non è in grado di giustificare. Ad esempio, la molecola dell’Ossigeno O2 risulta essere paramagnetica e ciò è compatibile solo con la presenza al suo interno di elettroni spaiati che, ne’ la teoria di Lewis, ne’ la teoria VB è in grado di giustificare.
La teoria degli orbitali molecolari considera la molecola come un insieme di nuclei e di elettroni e, valutando le loro reciproche interazioni, determina le funzioni d’onda che descrivono gli elettroni nella molecola in modo analogo a quello usato per individuare le funzioni d’onda che descrivono gli elettroni negli atomi isolati.
Gli elettroni di una molecola vengono descritti da funzioni d’onda dette orbitali molecolari le cui superfici limite si estendono su tutta la molecola. Le superfici limitedegli orbitali molecolari sono policentriche, abbracciando tutti i nuclei della molecola, a differenza di quelle degli orbitali atomici (OA) che sono monocentriche, ovvero riferite ad un solo nucleo. In altre parole tutti gli elettroni della molecola risentono dell’attrazione di tutti i nuclei e ciascun elettrone contribuisce a tenere insieme tutta la molecola.
La teoria MO prevede che, quando due atomi si legano, tutti i loro orbitali atomici (AO) di valenza si combinino per dare altrettanti orbitali molecolari (MO). La molecola più semplice è quella di H2+, costituita da un elettrone sottoposto all’azione di due protoni posti ad una certa distanza l’uno dall’altro. In questo caso si può risolvere l’equazione di Schrödinger in modo rigoroso e trovare le funzioni orbitali e i valori delle energie. In tutti gli altri casi (sistemi a più elettroni) non è possibile risolvere l’equazione d’onda ed è pertanto necessario ricorrere a metodi approssimati che tengano conto in qualche modo delle interazioni interelettroniche.
Il metodo di approssimazione più semplice e normalmente utilizzato è noto come L.C.A.O. (Linear Combination of Atomic Orbitals), in cui le funzioni d’onda degli orbitali molecolari si ottengono come combinazione lineare delle funzioni d’onda degli orbitali atomici.
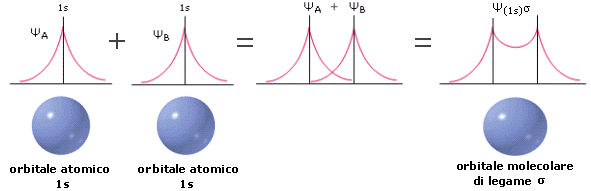
Attraverso il metodo L.C.A.O. le funzioni d’onda di due orbitali atomici si combinano per somma (inteferenza costruttiva) e per sottrazione (interferenza distruttiva) generando le funzioni d’onda di altrettanti orbitali molecolari.
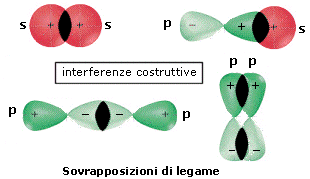
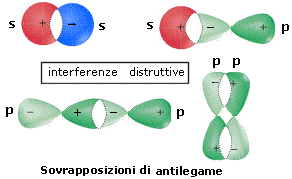

Come avviene negli orbitali atomici, anche negli orbitali molecolari la probabilità di trovare gli elettroni è data dal quadrato della funzione d’onda Ψ2. Se, ad esempio. combiniamo due orbitali atomici con funzioni d’onda ψA e ψB, otterremo
orbitale molecolare di legame
Ψ = ψA + ψB
Ψ2 = (ψA + ψB)2 = ψA2 + ψB2 + 2ψAψB
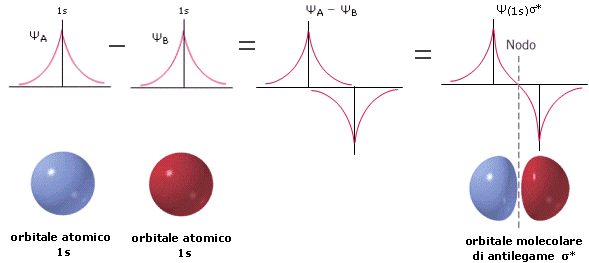
orbitale molecolare di antilegame
Ψ* = ψA - ψB
(Ψ*)2 = (ψA - ψB)2 = ψA2 + ψB2 - 2ψAψB
Come si può osservare, la probabilità di trovare l’elettrone in un orbitale molecolare differisce dalla semplice somma delle probabilità di trovare l’elettrone nei due orbitali atomici (ψA2 + ψB2) per il termine 2ψAψB. Tale termine, detto integrale di sovrapposizione, è positivo per gli orbitali molecolari di legame (nei quali dunque la probabilità di trovare l’elettrone è maggiore rispetto agli orbitali atomici separati). è negativo per gli orbitali molecolari di antilegame (nei quali dunque la probabilità di trovare l’elettrone è minore rispetto agli orbitali atomici separati) ed è nullo per gli orbitali molecolari di non legame (nei quali dunque la probabilità di trovare l’elettrone è uguale a quella degli orbitali atomici separati).
In generale se si combinano n orbitali atomici si ottengono n orbitali molecolari, metà di legame e metà di antilegame. Quando, dalla combinazione degli orbitali atomici, si genera un numero dispari (2n+1) di orbitali molecolari, allora n sono orbitali di legame, n sono orbitali di antilegame e 1 è un orbitale di non legame.
L’energia degli orbitali molecolari è correlata al numero di nodi presenti. L’orbitale di legame a più bassa energia non presenta nodi. Maggiore è il numero dei nodi, maggiore è l’energia dell’orbitale molecolare.
Affinché due o più orbitali atomici si possano combinare linearmente fra loro per formare orbitali molecolari devono essere soddisfatti i seguenti criteri:
1. Si possono combinare solo orbitali che possiedono energie non troppo diverse tra loro.
2. Le superfici di inviluppo degli orbitali atomici devono sovrapporsi il più possibile. Se due orbitali atomici hanno un’estensione limitata ed alla distanza di legame danno una sovrapposizione trascurabile (orbitali più interni) non possono formare orbitali molecolari. In altre parole, anche per la teoria MO vale la regola generale che ai legami contribuiscono essenzialmente gli orbitali più esterni (elettroni di valenza)
3. Si possono combinare solo gli orbitali che presentano la stessa simmetria rispetto all’asse internucleare. Tipicamente una sovrapposizione asimmetrica degli orbitali genera orbitali di non legame
Una volta costruiti tutti gli orbitali molecolari, questi vengono diagrammati insieme agli orbitali atomici genitori per visualizzare l'ordine crescente dell'energia che compete loro ed infine riempiti con tutti gli elettroni degli orbitali atomici che li hanno generati, seguendo le normali regole di aufbau.
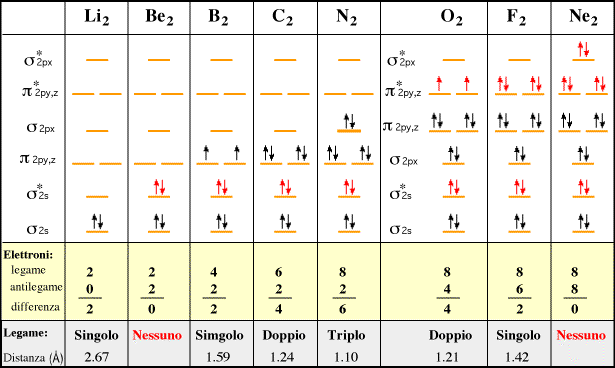
Il legame di una molecola è tanto più forte quanto maggiore è il numero di elettroni negli orbitali di legame rispetto al numero di elettroni negli orbitali di antilegame.
In generale si formerà un legame, e quindi una molecola, quando il numero di elettroni negli OM di legame (ne) supera il numero di elettroni negli OM di antilegame (ne*).
Si definisce ordine di legamela metà della differenza tra il numero degli elettroni negli orbitali di legame e il numero degli elettroni negli orbitali di antilegame (gli elettroni negli eventuali orbitali molecolari di non legame non contribuiscono).
OL = (ne - ne*)/2
Quanto più elevato è l’ordine di legame, tanto minore è la distanza internucleare e tanto maggiore è l’energia di legame.
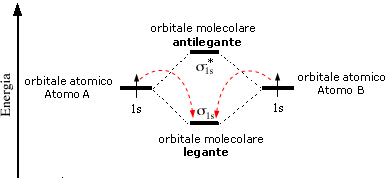
Vediamo ad esempio il metodo MO applicato alla molecola biatomica dell’Idrogeno H2.
Se indichiamo i due atomi di Idrogeno che si legano con HA e HB, le due funzioni d’onda che si sommano e si sottraggono per dare i due orbitali molecolari sono ΨA(1s) e ΨB(1s).
Prima sommiamo le due funzioni d’onda degli orbitali atomici 1s, ottenendo la funzione d’onda dell’orbitale molecolare di legame Ψσ1s. In questo caso il valore di Ψ (e quindi anche di Ψ2) aumenta nella regione tra i due nuclei. L’aumentata densità elettronica internucleare (maggior probabilità di trovare l’elettrone) scherma le cariche positive nucleari e genera una forza attrattiva sui due nuclei che li tiene legati (OM di legame).
Poi sottraiamo le due funzioni d’onda degli orbitali atomici 1s, ottenendo la funzione d’onda dell’orbitale molecolare di antilegame Ψσ*1s, la quale presenta un piano nodale passante tra i due nuclei atomici. In questo caso il valore di Ψ (e quindi anche di Ψ2) diminuisce fino ad annullarsi nella regione tra i due nuclei. La diminuita densità elettronica internucleare (minor probabilità di trovare l’elettrone) non è in grado di schermare le cariche positive nucleari e di generare una forza attrattiva sui due nuclei (OM di antilegame).
Gli orbitali atomici di partenza ed i due orbitali molecolari ottenuti vengono riportati in un diagramma in funzione del loro contenuto energetico. Come abbiamo già detto l’orbitale molecolare di legame è più stabile degli orbitali atomici di partenza, mentre quello di antilegame è meno stabile. i due elettroni inizialmente presenti negli orbitali atomici di partenza si sistemano dunque nell’orbitale molecolare a più bassa energia (Principio di minima energia) con spin antiparallelo (principio di Pauli) che risulta essere l’orbitale di legame. Il diagramma suggerisce che l'energia della molecola è minore rispetto a quella associata ai due atomi isolati risultando pertanto un sistema più stabile.
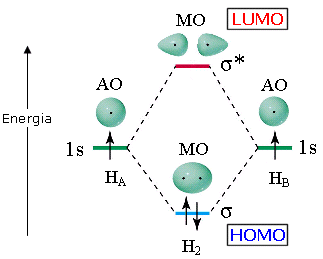
L’ultimo orbitale molecolare contenente elettroni è detto HOMO (Highest Occupied Molecular Orbital). Il primo orbitale molecolare vuoto è detto LUMO (Lowest Unoccupied Molecular Orbital). HOMO e LUMO sono definiti orbitali molecolari di frontiera.
L’andamento dell’energia potenziale per i due orbitali molecolari della molecola dell’Idrogeno in funzione della distanza interatomica è la seguente
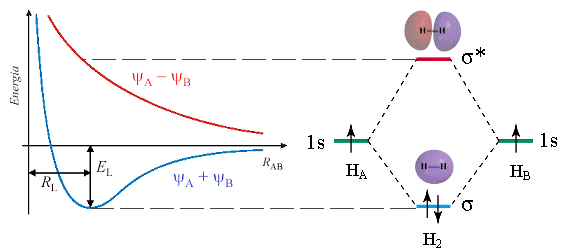
Elettroni in orbitali di legame ne = 2
Elettroni in orbitali di antilegame ne* = 0
Ordine di legame = (ne - ne*)/2 = (2 – 0) / 2 = 1.
La molecola biatomica dell’idrogeno è tenuta insieme da un legame covalente semplice.
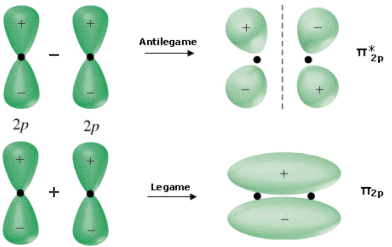
Gli orbitali p si possono combinare tra loro in due modi: frontalmente, generando orbitali molecolari σ e σ*

oppure lateralmente, generando orbitali π e π*.
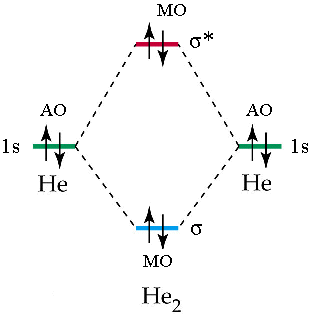
La teoria MO può spiegare perché certi composti non si formano. Se andiamo. ad esempio a diagrammare i livelli energetici della ipotetica molecola di He2, troviamo che i due elettroni nell’orbitale antilegante annullano l’effetto dei due elettroni nell’orbitale legante. L’ordine di legame è (2 – 2)/2 = 0 (nessun legame).
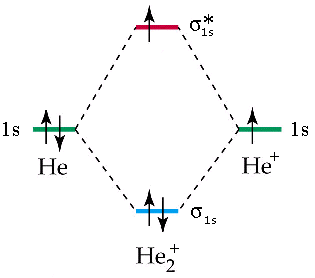
Mentre esiste lo ione He2+ con un ordine di legame pari a (2 – 1)/2 = 0,5
La teoria MO può spiegare le proprietà paramagnetiche della molecola dell’Ossigeno. Se andiamo a diagrammare i livelli energetici della molecola di O2, troviamo che negli orbitali antileganti a più alta energia vi sono due elettroni spaiati (regola di Hund) che giustificano il fenomeno del paramagnetismo osservato sperimentalmente e non interpretabile con la teoria VB. Le molecole paramagnetiche manifestano un momento magnetico intrinseco, ma a causa dell'agitazione termica il momento magnetico medio è nullo, tuttavia sotto l'azione di un campo magnetico esterno si verifica un fenomeno di parziale orientazione delle molecole con la comparsa di un momento magnetico risultante concorde al campo esterno (paramagnetismo).
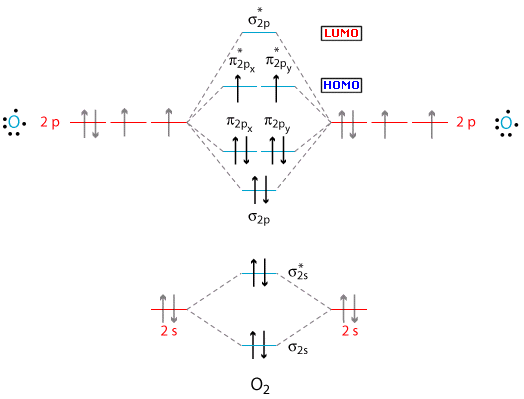
La configurazione della molecola dell’ossigeno è
(σ2s)2 (σ2s*)2 (σ2p)2 (π2p)4 (π2p*)2
ed il suo ordine di legame è OL = ½ (2 - 2 + 2 + 4 – 2) = 2 = Legame doppio
Fonte: http://www.pianetachimica.it/didattica/documenti/Chimica_Generale.doc
Sito web da visitare: http://www.pianetachimica.it
Autore del testo: non indicato nel documento di origine
Il testo è di proprietà dei rispettivi autori che ringraziamo per l'opportunità che ci danno di far conoscere gratuitamente i loro testi per finalità illustrative e didattiche. Se siete gli autori del testo e siete interessati a richiedere la rimozione del testo o l'inserimento di altre informazioni inviateci un e-mail dopo le opportune verifiche soddisferemo la vostra richiesta nel più breve tempo possibile.
I riassunti , gli appunti i testi contenuti nel nostro sito sono messi a disposizione gratuitamente con finalità illustrative didattiche, scientifiche, a carattere sociale, civile e culturale a tutti i possibili interessati secondo il concetto del fair use e con l' obiettivo del rispetto della direttiva europea 2001/29/CE e dell' art. 70 della legge 633/1941 sul diritto d'autore
Le informazioni di medicina e salute contenute nel sito sono di natura generale ed a scopo puramente divulgativo e per questo motivo non possono sostituire in alcun caso il consiglio di un medico (ovvero un soggetto abilitato legalmente alla professione).
"Ciò che sappiamo è una goccia, ciò che ignoriamo un oceano!" Isaac Newton. Essendo impossibile tenere a mente l'enorme quantità di informazioni, l'importante è sapere dove ritrovare l'informazione quando questa serve. U. Eco
www.riassuntini.com dove ritrovare l'informazione quando questa serve