I riassunti , gli appunti i testi contenuti nel nostro sito sono messi a disposizione gratuitamente con finalità illustrative didattiche, scientifiche, a carattere sociale, civile e culturale a tutti i possibili interessati secondo il concetto del fair use e con l' obiettivo del rispetto della direttiva europea 2001/29/CE e dell' art. 70 della legge 633/1941 sul diritto d'autore
Le informazioni di medicina e salute contenute nel sito sono di natura generale ed a scopo puramente divulgativo e per questo motivo non possono sostituire in alcun caso il consiglio di un medico (ovvero un soggetto abilitato legalmente alla professione).
CORSO DI CHIMICA GENERALE
La chimica è la scienza che studia le proprietà, la composizione e le trasformazioni della materia.
Una trasformazione è il passaggio fondamentale dello studio della chimica:
La chimica ha inizio con la legge di Lavoisier (della massa costante), la quale afferma che
Una reazione può richiedere una determinata energia o calore d‘innesto, e ne può liberare durante la reazione.
La chimica descrive in modo oggettivo queste trasformazioni.
Lo strumento principe che descrive le proprietà degli elementi è la tavola periodica, in cui gli elementi sono organizzati secondo determinate proprietà.
Aristotele fu il creatore di quel dogma che è durato per secoli.
Affermò che il mondo può essere spiegato con 4 elementi (acqua, aria, terra, fuoco)che mescolandosi in diverse proporzioni formano le diverse proprietà della materia, come i colori.
Le qualità caldo, secco, freddo e umido sono la chiave per le trasformazioni.
Questa impostazione è rimasta sostanzialmente inalterata fino a Lavoisier
I metalli erano sette, tutti provenienti da terre differenti che, bruciate con il carbone, avrebbero dato il metallo puro.
Oro e rame erano i più puri, quasi divini, poiché si trovavano in natura, mentre gli altri necessitavano di esser estratti e raffinati.
Varie credenze, come quelle di Paracelso e Becher cercarono di andare oltre la chimica de 4 elementi aristotelici.
La pubblicazione del fisico e chimico Boyle, Sceptical chymist, fu un inizio di messa in discussione radicale della teoria dei 4 elementi, che verrà messa in disparte definitivamente da Lavoisier.
Lavoisier scoprì con esperimenti in cui metteva animali e una candela sotto una campana di vetro, che l’aria era composta da due differenti elementi:
Anche l’acqua è composta da due elementi, ma non è una miscela, bensì un composto di ossigeno ed idrogeno.
Con queste scoperte, Lavoisier instaura la crisi della chimica dei 4 elementi.
Un’aria fissa è liberata dai carbonati, ed è simile a quella del nostro respiro (CO2). E’ mefitica.
4/5 dell’aria è mefitica, 1/5 è buona.
Sono sostanze che non possono essere più scomposte chimicamente.
Lavoisier ne indicò 33 e tra queste nominò:
Oggi sono circa 100 gli elementi e molti non rispecchiano la catalogazione di Lavoisier
Vi furono differenti modi di proporre la classificazione degli elementi.
Mendelev fu colui che organizzò gli elementi in una tavola periodica, in ordine crescente secondo il numero di massa, e disposti in colonne a seconda di determinate proprietà fisico-chimiche.
La tavola periodica degli elementi è attualmente aggiornata con tutti gli elementi stabili che sono sulla terra. È definitiva.
Teoria formulata da John Dalton nel 1803:
La scoperta della chimica porta con sé come conseguenza anche quella della crisi del vitalismo:
La materia è tutto ciò che possiede una massa ed occupa un volume.
La materi è costituita da atomi, i quali sono formati da tre particelle subatomiche:
Si definisce peso atomico di un elemento la massa relativa e media di quell'elemento rispetto ad 1/12 della massa di un nuclide di 12C.
Il peso atomico relativo (PA) pari l rapporto tra la massa assoluta dell’atomo e l’unità di massa atomica (in kg).
Il peso molecolare (PM) è uguale alla somma dei pesi atomici, ognuno moltiplicato per il proprio indice.
Una mole rappresenta la quantità di un elemento il cui peso, espresso in grammi, è pari al suo peso atomico o molecolare.
Una mole di qualsiasi sostanza contiene NA = 6,027 1023 particelle.
Per calcolare il numero di moli di una massa di sostanza si rapporta la massa (g) della sostanza al peso molare (g/mol).
Le sostanze che sono formate da più elementi sono dette composti chimici, o semplicemente composti.
Una molecola è la più piccola particella di un composto che ne possiede tutte le proprietà chimiche. È un aggregato discreto di atomi tenuto insieme da legami chimici.
Un composto è determinato da una legge detta della composizione costante:
Le proprietà di un composto sono differenti da quelle degli elementi che ne formano le molecole.
La materia può presentarsi sotto forma di:
vi possono essere due differenti tipi di miscugli:
le proprietà fisiche sono una composizione di quella degli elementi che compongono la miscela.
Una miscela è tale se è possibile separare i vari composti che la compongono mediante procedimenti fisici:
Vi sono differenti tipi di formule:
Alcune molecole non sono discrete quindi vengono rappresentate mediante formule minime (es. NaCl, CaCl2, Fe, O, ecc…)
In alcuni casi possono esservi dei composti formati con ioni la cui carica elettrica risultante sia nulla:
Per ricavare la formula vera dai pesi dei vari componenti si seguono i seguenti passaggi:
Si considera la materia come un insieme di particelle.
Queste particelle sono sottoposte a delle forze di interazione che le legano l’una all’altra. A seconda della forza di interazione tra molecole si individuano tre stati fisici:
Il passaggio di stato di una sostanza avviene per perdita o acquisizione d’energia cinetica da parte delle particelle. Questo permette una variazione della temperatura.
I passaggi di stato sono così chiamati:
Nei primi tre la materia acquista energia, negli ultimi tre la cede.
La pressione di vapore è la pressione del vapore in un recipiente chiuso in equilibrio con un liquido (quando non evapora più nulla). È una sorta di equilibrio dinamico, in cui vi è continuo scambio tra il liquido ed il vapore.
Energia di passaggio di stato
Quando in un liquido una molecola ha una certa energia che supera una energia critica, questa risale in superficie ed evapora.
Il numero delle molecole che si trovano al di sopra dell’energia critica è dato dalla legge NE = N e-E/RT.
Questa equazione da luogo ad una campana in un grafico statistico del tipo (n° x E).
Si noti che la pressione di vapore varia a seconda della temperatura in k secondo la legge Pv = k N e-E/RT.
A 100°C e 1 atm in un recipiente aperto l’acqua bolle, poiché Pv=Patm.
Durante i passaggi di stato, la temperatura resta costante ed occorre una determinata energia per portarli a termine.
Non è solamente l’innalzamento della temperatura che può permettere un passaggio di stato, ma anche la variazione della pressione o del volume:
Per quanto riguarda la sublimazione (da solido ad aeriforme), la pressione di vapore del ghiaccio cresce con l’aumentare della temperatura.
Nei diagrammi di stato PxT, dell’acqua si può notare che in un solo punto, ovvero con pressione pari a 4,58 mmHg e a 0 gradi, si ha il punto triplo, in cui si può trovare l’acqua allo stato solido, liquido o di vapore.
La particolarità dell’acqua rispetto ad esempio alla CO2, è che la retta che descrive la pressione di fusione in funzione della temperatura ha un andamento decrescente, contrario a quello di tutti gli altri elementi.
La liquefazione dei gas
Quando un gas scende al di sotto del punto di ebollizione le molecole si avvicinano, agiscono le forze attrattive e il gas si liquefa.
Effetto Joule-Thomson
Quando in un gas si diminuisce la pressione, la maggior distanza tra le molecole causa una diminuzione della velocità media, quindi un raffreddamento.
Il calore è associato al movimento delle particelle:
In prossimità dei passaggi di stato, visono delle temperature critiche, alle quali per permettere il passaggio di stato è necessario fornire un maggior quantitativo di calore, che permetterà la trasformazione fisica e manterrà la temperatura costante finché il passaggio non sia completato.
A temperatura costante, vi sono anche delle pressioni critiche, che permettono il passaggio di stato.
Quando in un gas la temperatura scende sotto il punto di ebollizione:
Un gas è un insieme di molecole separate che si muovono in modo caotico.
È comprimibile e molto sensibile ai cambiamenti di temperatura.
Un gas è definito da 4 variabili:
La pressione
Si definisce come il rapporto tra una forza esercitata sull’unità di superficie: P= F/S.
La pressione di un gas sulle pareti di un recipiente è data dall’insieme delle collisioni delle molecole.
La pressione si misura in:
La temperatura
Rappresenta la intensità degli urti delle particelle, essendo strettamente legata alla loro velocità.
Si misura in:
Gli 0 K, o anche – 273,15°C, rappresentano lo zero assoluto ovvero una temperatura al di sotto della quale non può esistere la materia (cfr legge di Boyle, altrimenti avrebbero V negativo)
la legge di Avogadro
il volume di un campione di gas ad una data temperatura e pressione è proporzionale al numero n delle moli di molecole presenti nel campione, indipendentemente dalla natura chimica.
V= k n
In condizioni standard, una mole di gas occupa sempre circa 22,4 l.
Legge dei gas ideali
Unendo le leggi di Boyle, Charles, Avogadro, si ha un unica legge, approssimativamente valida per tutti i gas ideali:
PV=nRT
La costante R è detta costante dei gas, ed è universale. Vale circa:
La legge dei gas ideali è un’equazione di stato, poiché non rappresenta l’andamento, ma ne descrive lo stato.
La legge di Boyle
Questa legge regola le trasformazioni isoterme (T costante).
A temperatura costante, volume e pressione sono inversamente proporzionali.
PV=k
Le leggi di Gay-Lussac
Trasformazione isocora (V costante): P = P0 (1 + aT )
Trasformazione isobara (P costante): V = V0 (1 + aT )
Altre leggi
Con n e P costanti: V/V0=P/P0.
Con n e V costanti: T/T0=P/P0.
La densità di un gas
La massa di un campione si calcola moltiplicando la massa molecolare per il numero di moli:
m = nM
Poiché la densità è il rapporto tra la massa ed il volume
d = nM / V
poiché
n/V = P/ RT
si afferma che: d = P M / RT.
Si può dunque affermare che la densità di un gas è proporzionale alla massa molecolare.
Volume molare
Si definisce volume molare il volume che occupa una mole di gas.
Tutti i gas, per la legge di Avogadro, a pari condizioni (P, T, V) hanno il medesimo volume.
Il volume molare di un gas è sempre molto più grande di quello di un liquido o di un solido,con differenze di ordine di grandezza delle migliaia.
Miscele di gas.
Pressione parziale: in una miscela, la pressione parziale di un gas è la P che esso eserciterebbe nelle stesse condizioni se occupasse il medesimo contenitore.
Legge di Dalton (o dele pressioni parziali): la pressione totale di una miscela di gas è pari alla somma delle pressioni parziali dei vari gas che la compongono.
P1= n1 RT / V
Frazione molare: è il numero di moli di un gas rispetto al numero di moli di una miscela.
X1= n1 / ntot.
Somma (ni) = 1
L’equazione di stato per le miscele
Ptot=Pa+Pb= ntotRT / V
Facendo il rapporto membro a membro tra l’eq di stato di una P parziale e quella della P totale, si ottienila frazione molare.
Pa/Ptot=Xa
Un gas è un insieme di molecole istanti l’una dalle altre. La teoia cinetica ei gas si fonda su tre assunzioni:
La teoria cinetica dei gas afferma che il volume e la pressione sono in relazione mediante l’equazione:
1/3nMv2=nRT
Dove n è il numero di moli, M la massa molecolare, v la velocità quadratica media delle molecole.
Questa legge indica che la velocità dipende SOLO dalla temperatura. Lo zero assoluto non è raggiungibile perché tutte le molecole sarebbero ferme.
I gas reali
I composti in fase gassosa non sono mai gas perfetti, ma vi è sempreun volume delle particelle e delle interazioni tra le molecole.
Un gas reale si avvicina all’idealità quando:
In sostanza si avvicina all’idealità quando le sue molecole sono il più possibile distanti tra loro.
Facendo l’operazione inversa il gas si allontana dall’idealità e cambia di stato diventando liquido.
equazione di van der Waals per i gas reali
(P + a n2/V2)(V-nb) = nRT
Se aumentano le forze di attrazione diminuisce la pressione totale
a e’ diverso da gas a gas.
Il volume a disposizione delle particelle si ottiene sottraendo al volume del recipiente quello occupato dalle molecole
b e’ una costante tipica di ogni gas.
Distribuzione delle velocità molecolari secondo Maxwell
Ogni molecola va incontro a cambiamenti continui della propria velocità.
La campana che dercrive la percentuale di molecole che vnno ad una determinate velocità è stata individuata da Maxwell. Questa curva viene chiamata distribuzione.
Diffusione
La penetrazione di una sostanza in un’altra è chiamata diffusione.
Due gas si compenetrano tra loro perché le molecole sono in movimento e libere di muoversi:
Legge di Graham
La velocità di diffusione di un gas è inversamente proporzionale alla radice quadrata del peso molecolare delle sue molecole.
A T costante V1/V2= (M2/M1)1/2
La velocità delle molecole in un gas è simile a quella del suono, di fatti il suono si propaga grazie al movimento di molecole.
Una reazione chimica è una trasformazione della materia legata a:
una reazione chimica è definita mediante un’equazione chimica.
Una reazione DEVE sempre rappresentare un processo che avviene in realtà, poiché non può riflettere un processo che non avviene.
Equazioni chimiche
Una reazione è scritta con una formula al cui centro c’è una freccia, a sinistra gli elementi di partenza di una reazione, i reagenti, a destra gli elementi finali, i prodotti.
La freccia significa “produce”, il simbolo “+” indica che le molecole entrano insieme nella reazione.
La simbologia prevede:
In una reazione, i numeri che prepongono le molecole sono detti coefficienti stechiometrici, che indicano il numero di molecole che interagiscono.
Talvolta, si mettono anche le quantità di energia che intervengono in una reazione, come se fossero delle molecole.
Per il principio della conservazione delle masse (nulla si crea e nulla si distrugge) l’equazione chimica deve essere bilanciata:
Per bilanciare un’equazione chimica si osservano 4 passaggi base:
è lo studio degli aspetti quantitativi delle reazioni chimiche:
massa reagente / massa molare reagente = moli reagente
Moli prodotto = coeff. Stechiometrico/moli reagenti * moli reagente
moli prodotto * peso molecolare prodotto = massa prodotto
massa prodotto effettiva / massa prodotto teorica * 100% = resa %
Un reagente si definisce limitante quando non è in quantità sufficiente per determinare il consumo completo degli altri reagenti.
Gli altri reagenti sono in eccesso quando sono in quantità superflue rispetto ad altri reagenti in modo da non rendere possibile un consumo completo nel prodotto.
Vi sono circa 5 tipi di reazione:
Una ossido-riduzione è una reazione in cui dei reagenti si scambiano elettroni per formare dei prodotti.
numero di ossidazione: carica che un atomo assumerebbe se tutti gli elettroni impegnati in legame si trovassero sull’atomo più elettronegativo.
Per stabilire il numero di ossidazione è necessario tenere presente che:
Gli elementi non metalli possono assumere differenti valori di ossidazione, solitamente tendenti alla configurazione otteziale.
L’azoto assume tutti i numeri di ossidazione, da -3 a +5.
Per il bilanciamento delle reazioni di ossidoriduzione si seguono i seguenti passaggi:
Uno ione positivo (catione) viene sempre indicato con la parola ione seguita dall’elemento corrispondente. Ad esempio Na+ si denomina ione sodio.
Quando gli ioni formati da un elemento sono più di uno si segue la seguente tabella:
Nel caso degli ioni negativi (anioni), se sono monoatomici si fa seguire al nome dell’elemento il suffisso –uro.
Nel caso di OSSIANIONI si aggiunge all’elemento il suffisso –ato, nel caso in cui siano monoatomici.
Qualora possono esservi più ossianioni con lo stesso elemento si segue a nomenclatura della seguente tabella.
Ipo |
- ito |
|
- ito |
|
- ato |
per |
- ato |
Sono composti formati da idrogeno e un metallo. In questi composti l’idrogeno assume numero di ossidazione pari a -1.
La formula generale è MeHx.
La nomenclatura tradizionale li chiama: “idruro di X”.
Gli idracidi sono composti dell’idrogeno con i non metalli, prevalentemente gli alogeni. Formula generica è HxnonMex.
La nomenclatura li chiama “acido nonMe-idrico”.
Questi composti sono pochissimi e si possono riassumere in una tabella.
HF |
Acido fluoridrico |
HBr |
Acido bromidrico |
HCl |
Acido cloridrico |
HI |
Acido iodidrico |
HCN |
Acido cianidrico |
H2S |
Acido solfidrico |
Gli osidi basici sono composti formati dal legame di ossigeno con un metallo. La formula generale è Me2Ox.
La nomenclatura nel caso i due elementi abbino un solo numero di ossidazione a cui legarsi li chiama con la formula “ossido di Me”.
Se i numeri di ossidazione sono più d’uno si segue il seguente schema:
Ipo |
- oso |
|
- oso |
|
- ico |
per |
- ico |
Le anidridi sono composti formati dal legame di ossigeno con un non metallo, con formula generale nMe2Ox.
Se vi è un solo numero di ossidazione compatibile si nominano “anidride nMe -ica”.
Se i numeri di ossidazione sono più d’uno si segue il seguente schema:
Ipo |
- oso |
|
- oso |
|
- ico |
per |
- ico |
Sono i derivati delle anidridi per aggiunta di una o più molecole d’acqua.
La formula generale è HxnMeOy.
La nomenclatura segue quella delle anidridi corrispondenti, ma si sostituisce la parola anidride con acido, formando nomi così strutturati “acido X-ico”.
Con l’aggiunta di più molecole d’acqua, si aggiungono i prefissi meta (1 H2O), piro- (2 H2O), orto- (3 H2O).
Sono composti derivati dall’unione di un metallo con uno o più gruppi idrossile, con formula generale Me(OH)x.
Si nomenclano con “idrossido di Me” e nel caso vi possano essere più numeri di ossidazione si segue lo schema:
Ipo |
- oso |
|
- oso |
|
- ico |
per |
- ico |
Derivano dalla sostituzione di uno o più idrogeni da un idracido o da un ossiacido, con un metallo.
La formula generale diventa Mex(RA)y.
La nomenclatura segue quella dall’acido da cui deriva, con le seguenti desinenze (pari a quelle degli ioni):
Ipo |
- ito |
|
- ito |
|
- ato |
per |
- ato |
Secondo la teoria atomica, che si fa risalire a Dalton, un atomo è una particella, la più piccola particella dotata di caratteristiche chimiche dell’elemento che rappresenta.
La teoria si articola nei seguenti punti:
Un elemento è una sostanza che è costituita da un solo tipo di atomi, ed ha proprietà fisiche che dipendono dal comportamento dell’insieme degli atomi di tale sostanza.
Thompson è stato il primo a determinare il rapporto carica-massa dell’elettrone. La carica è stata poi misurata da millikan.
Gli elettroni sono in uno spazio intorno al nucleo
In un atomo elettricamente neutro il loro numero è pari a quello de protoni.
La carica di un eletrone è – 1,602 10-19 C.
Ha una massa molto piccola, quasi trascurabile (9,1 1028 g)
Rutherford ha mostrato che il rapporto massa/carica differiva con il tipo di gas usato.
Il rapporto più alto era ottenuto con l’idrogeno.
Il protone (H+) ha carica uguale ed opposta a quella dell’elettrone e massa molto più grande (1,67 10-24).
Ha massa molto simile a quella del protone, ma carica neutra.
Solitamente il numero dei protoni e neutroni è uguale. Quando differisce si parla di isotopi.
Maxwell nel 1864 sviluppa la teoria delle onde elettromagnetiche, arrivando all’equazione c = ln
Le onde elettromagnetiche sono onde oscillanti perpendicolarmente tra i due campi: elettrico e magnetico.
Negli spettri elettromagnetici si riesce ad osservare le lastre delle onde luminosa, con lunghezze d’onda tali per cui sono visibili.
Le onde ad alta energia hanno una lunghezza d’onda molto piccola, quelle a bassa energia una molto grande.
Plank ipotizzò che le emissioni di un corpo riscaldato emettessero radiazioni:
In definitiva scopre che l’energia emessa è:
Balmer e Rydberg trovarono le equazioni che descrivevano le lunghezze d’onda emesse dall’idrogeno:
Si giunge quindi al modello atomico di Niels Bohr, che prevede:
Nel 1924 il fisico e chimico De Broglie suggerisce che anche l’elettrone deve avere proprietà ondulatoria:
da ciò scaturisce il principio di indeterminazione di Heisenberg:
“non è possibile definire esattamente la posizione di un elettrone se questo è in movimento”.
Sebbene l’elettrone abbia natura una velocità, dovrebbero essere attratti dal nucleo.
In più, il movimento comporta l’emissione di radiazioni elettromagnetiche, quindi la perdita dell’energia:
L’energia degli elettrico risulta essere quantizzata:
Nell’equazione di Schroedinger un elettrone è definito da quattro numeri quantici:
Se due elettroni che ruotano nello stesso verso hanno spin parallelo, due che ruotano in senso opposto sono in spin opposto.
Per determinare quali orbitali vengono occupati s applicano le regole dell’aufbau, cioè il modo di riempimento convenzionale degli orbitali.
La particolarità del riempimento si ha con Z = 19, per cui non si occupa come si dovrebbe l’orbitale 3d, ma il 4s, che ha un livello energetico minore. L’orbitale 3d si inizia a riempire dallo scandio (Z=21).
L’aufbau permette di prevedere con una discreta correttezza l’80% delle configurazioni elettroniche. Ci sono delle eccezioni:
Naturalmente anche il riempimento degli elettroni di ioni segue queste regole.
La configurazione del livello più esterno degli elementi determina la loro posizione nella tavola periodica:
La tabella periodica degli elementi è stata proposta da Mendelev nel 1869, che dispose gli elementi in ordine di peso atomico in gruppi e in periodi.
La tabella è divisa in blocchi corrispondenti alla configurazione elettronica esterna:
I periodi invece sono numerati a seconda del numero quantico principale n: ad ogni gruppo corrisponde un livello energetico.
Il fatto che le configurazioni elettroniche esterne siano identiche in ogni gruppo conferisce agli elementi di ogni gruppo caratteristiche chimiche simili, che si articolano in maniera periodica lungo la tabella.
Il raggio atomico dipende dagli elettroni presenti, e dal numero di elettroni presenti nell’ultimo livello energetico:
Nel caso dei raggi degli ioni:
Per quanto riguarda l’energia di ionizzazione, aumenta con l’aumentare degli elettroni di valenza (da sx a dx) e diminuisce con l’aumentare del raggio atomico (dall’alto verso il basso).
Le basse energie di ionizzazione a sinistra giustificano il carattere metallico:
gli elementi del gruppo p sono caratterizzati da elettroni di valenza s con un elevatissima energia di ionizzazione. Sono detti per ciò doppietti inerti.
Si definisce affinità elettronica l’energia che si ottiene nel mettere un elettrone nell’orbitale ad energia minore.
Esprime la tendenza dell’atomo a ricevere elettroni. Questa tendenza si ha dal terzo al settimo gruppo della tavola, aumentando verso sinistra:
Sono definiti metalli gli elementi che tendono a perdere elettroni con facilità nei loro composti di combinazione, a causa di:
gli elementi del primo, del secondo e del terzo gruppo sono detti metalli, per la loro tendenza a formare cationi.
Gli elementi del V, VI, VII gruppo sono detti non metalli, per la loro tendenza a formare legami covalenti o anioni.
Gli elementi del III e del IV gruppo hanno caratteristiche intermedie e sono definiti anfoteri, poiché presentano caratteristiche intermedie a seconda dei legami che formano, tra metalli e non metalli.
La chimica nucleare studia la struttura e le trasformazioni che avvengono a livello del nucleo atomico.
Avviene che i nuclei cambiano la propria struttura atomica spontaneamente e nel farlo emettono radiazioni.
Il nucleo, nel modello più semplice universalmente accettato, è un insieme di nucleoni:
la forza di attrazione che avviene con dispendio di massa (convertita in energia secondo la formula E=mc2 ) tra i protoni nel nucleo vince le forze di attrazione che si sviluppano all’interno dello stesso.
Solitamente il nucleo di un atomo è stabile. Superati gli 83 protoni il nucleo inizia a diventare instabile e va incontro a trasformazioni.
Un elemento è caratterizzato dal numero di protoni, ma può differire per la presenza di neutroni:
Per i primi venti elementi della tavola periodica il numero di protoni è uguale al numero di neutroni.
Quando gli elementi hanno Z>21 il rapporto neutroni/protoni è maggiore di 1 ed ha un incremento continuo.
Da elementi con Z>83, si inizia ad avere elementi instabili:
I nuclei stabili, sono pochissimi rispetto alle combinazioni possibili tra i vari nucleoni:
un decadimento radioattivo è un processo spontaneo in cui il nucleo cambia la propria struttura:
è l’emissione di un nucleo di elio, ovvero una molecola di He2+.
L’atomo perde due protoni e due neutroni (Z=-2; A=-4).
Il decadimento beta comporta l’emissione di una particella b- (un elettrone), con la trasformazione di un neutrone in un protone.
I numeri cambiano:
é la semplice emissione di un fotone ad alta energia, accompagna tutti i decadimenti.
Un nucleo cattura uno dei propri elettroni e trasforma un protone in un neutrone (Z-1).
Avviene con la trasformazione del Calcio-41 in potassio (K-41)
Un protone si trasforma in un neutrone e viene emesso un positrone (massa identica all’elettrone con carica +1).
Il numero atomico diminuisce di una unità (Z-1).
A seconda se un isotopo si colloca al di sopra o al di sotto della banda di stabilità possono avvenire differenti tipi di decadimento:
Quando vi sono nuclidi pesanti e particolarmente instabili (sopra Z=83) si hanno i decadimenti alfa, con emissione di 4 nucleoni (2 neut e 2 prot)
Gli elementi tendono sempre ad assumere delle configurazioni stabili, quindi vi sono delle proprie serie radioattive tracciabili e confrontabili a partire da determinati isotopi.
Quando il nucleo emette una radiazione (a, b+ o b-) il numero atomico varia e si forma un nuovo elemento.
La velocità con cui avvengono queste emissioni può essere misurata contando le radiazioni emesse nell’unità di tempo:
Esiste una legge di proporzionalità diretta tra la velocità con cui un elemento decade (A, attività dell’elemento) e il numero di decadimenti (N), espressa attraverso una costante (k, costante di decadimento dell’isotopo):
A=kN
Un’altra legge esprime questa velocità: ln (N/N0) = -kt.
In base a questa legge si definisce l’emivita il tempo necessario affinché decada la metà degli atomi dell’isotopo radioattivo:
ln(N/2N)=-kt1/2
ln(1/2)=-kt1/2
t1/2= 0,963/k
Il tempo di dimezzamento (emivita) di un isotopo radioattivo dipende dalla costante di decadimento, la quale dipende dalle caratteristiche dell’isotopo.
Per la datazione dei reperti viene utilizzato un isotopo del carbonio, il C-14, che ha un’emivita di 5730 anni:
La radioattività possiede differenti unità di misura:
In biologia e medicina, gli isotopi radioattivi possono essere usati come:
Per poter essere compatibili e non dannosi agli organismi devono avere:
Gli atomi interagiscono tra loro formando molecole. L’union di atomi prende il nome di legame e può avvenire in vari modi.
La teoria dei legami degli atomi si deve a Lewis:
Un legame covalente è formato da una coppia di elettroni messi in compartecipazione tra due atomi.
Possono verificarsi due casi:
Un legame covalente si ha tra due atomi del gruppo p della tavola periodica, oppure con H e Be.
Generalmente si osserva la differenza di elettronegatività tra i due atomi che formano un legame:
L’energia del legame covalente equivale all’energia necessaria per scinderlo.
La lunghezza di legame è definita come la distanza tra i nuclei dei due elementi. Dipende da:
L’entalpia di legame è l’energia che si libera quando il legame viene rotto.
A seconda della differenza di eletronegatività tra gli atomi considerati, un legame covalente può essere:
Un legame polare è un legame in cui, per la differenza di elettronegatività tra i due atomi, gli elettroni tendono a spostarsi verso uno dei due atomi, creando dei momenti dipolari differenti all’interno della stesa molecola:
Il legame ionico è l’attrazione elettrostatica tra le cariche di un anione e un catione:
Il solido ionico è disposto in modo tale che tra gli ioni si abbia un minimo di energia.
Un legame ionico è favorito da:
Gli atomi tendono il più possibile a completare i propri ottetti di elettroni nel livello energetico più esterno, lo fanno mediante coppie di elettroni compartecipate.
Questa regola funge benissimo con gli elettroni del secondo periodo (O, C, F, N, ecc..)
Quando entrano in giuoco anche gli orbitali d allora possono essere riempiti più orbitali, e in un atomo possono essere sistemati più di otto elettroni.
Per parecchie molecole, è possibile scrivere più di una formula di Lewis, sono le varie strutture limite, dette ibridi di risonanza.
Gli elementi dal terzo periodo hanno a disposizione gli orbitali d, di energia accessibile:
Un radicale è una porzione scissa da una molecola che ha almeno un elettrone spaiato (esempio radicale ossidrile, OH):
Spesso la struttura tridimensionale conferisce alla molecola le sue proprietà chimiche. La struttura tridimensionale può essere descritta mediante:
La struttura di Lewis fornisce indicazioni circa:
Tuttavia non fornisce informazione alcuna circa la struttura tridimensionale della molecola.
La forma di una molecola può essere dedotta seguendo i principi della teoria VSEPR, ovvero repulsione tra le coppie degli elettroni del livello di valenza:
Se è presente un atomo centrale allora gli atomi legati ad esso tendono a disporsi sulla superficie di una sfera alla massima distanza possibile.
La forma e l’angolo dipendono solamente dal numero dei sostituenti:
Tuttavia i doppietti solitari (lone pairs) fanno sentire il loro effetto:
I principi della teoria Vsepr possono essere applicati anche a molecole più complesse:
Una molecola polare è una molecola che possiede un momento dipolare differente da zero.
Una molecola che contiene legami non necessariamente è essa stessa polare:
Il momento dipolare m si calcola come m=Qr, dove Q è la carica e r la distanza tra i due poli.
In molecole tridimensionali, la somma dei vettori del momento dipolare di ciascun legame da la polarità della molecola.
Per gli elementi del secondo periodo occorre tenere presente che :
Per i legami occorre tener presente alcune regole:
Il legame covalente avviene solamente quando due orbitali si sovrappongono:
Gli orbitali possono ottenere una ibridazione mediante una cessione di poca energia:
L’ammoniaca potrebbe formare tre legami planari sui 3 orbitali 2p, invece ibrida gli orbitali e forma una struttura tetraedrica, con orbitali ibridati sp3:
Alcune geometrie molecolari che coinvolgono atomi centrali con ottetti espansi possono essere interpretati come l’ibridazione di orbitali ibridi sp con orbitali d per formare ibridi spd.
Lo stato di ibridazione lo si può dedurre dalla geometria molecolare in relazione alla presenza di doppietti liberi.
All’interno della teoria dell’orbitale di legame si possono anche spiegare i doppi e tripli legami:
Un orbitale molecolare è un orbitale esteso a due o più atomi uniti da legami covalenti:
Per approssimazione, si possono calcolare i moti di un elettrone intorno ad un nucleo attraverso la combinazione lineare degli orbitali atomici (metodo LCAO).
Con il metodo LCAO si possono individuare:
Un orbitale di legame risulta la somma delle funzioni d’onda dei due elettroni di legame:
![]()
Un orbitale di antilegame risulta essere definito dalla sottrazione dele funzioni d’onda dei due elettroni
![]()
Negli elementi più piccoli Del blocco p nel secondo periodo è possibile che si verifichi l’interazione tra orbitali s e p, con conseguente ibridazione:
L’ordine di legame è dato dal numero netto di legami che si ottiene dopo aver annullato legami con antilegami.
La formula che descrive l’ordine di legama (bond order) è:
![]()
Chiaramente se il bond order è nullo, la molecola (o il legame descritto) non esiste.
Le varie molecole sono attratte da forze intermolecolari che determinano l’aggregazione in liquidi e solidi.
Questi legami attrattivi tra molecole hanno un’energia molto minore a quelle dei legami covalenti (< 50 kJ/mol).
E’ il vero e proprio legame ionico, molto forte e di natura elettrostatica:
L’energia di un legame ionico è data dalla relazione: ![]() .
.
E’ l’interazione che si viene a formare quando sono disciolti in acqua gli ioni:
È responsabile dell’idratazione dei cationi in soluzione, ovvero del legame di una molecola d’acqua ad un catione centrale
L’energia di questa interazione è maggiore con cationi molto piccoli, poiché diminuisce con la distanza, secondo la legge: ![]()
Interazione di natura elettrostatica che si forma tra molecole polari, in cui vi è l’attrazione dei vari dipoli elettrici:
Infatti, l’interazione diminuisce secondo le leggi:
In una miscela di liquidi, uno polare e uno non polare si può assistere all’induzione di un dipolo:
Sono dovute alla formazione di dipoli istantanei durante il moto degli elettroni:
L’induzione di dipoli istantanei e delle conseguenti interazione è molto più frequente nelle molecole cilindriche piuttosto che in quelle sferiche.
L’energia delle forze di dispersione dipende dalla polarizzabilità(a):![]()
Il legae idrogeno è un legame di natura fortemente polare che avviene quando l’idrogeno si pone a ponte tra atomi fortemente elettronegativi.
È il responsabile delle alte temperature d’ebollizione dei composti dell’idrogeno con atomi fortemente elettronegativi (ad esempio l’acqua).
Il legame H è molto piccolo e presenta particolari caratteristiche, rispetto a tutti gli altri legami dipolo-dipolo:
Allo stato solido la molecola d’acqua è coinvolta in 4 legami H (2 H si legano a 2 O di altre molecole, 2 O accettano 2 legami H).
Allo stato liquido questi legami sono circa tre.
Le forti attrazioni che avvengono a livello dell’H2O sono la causa della repulsione e dell’espulsione di una molecola idrofoba:
Il legame ad idrogeno è il legame più forte, seguono le interazoni tra le altre molecole polari, poi tra quelle apolari.
I punti di ebollizione e fusione dipendono in larghissima misura dalle interazioni intermolecolari:
Sono definite soluzioni delle miscele omogenee in cui una sostanza (soluto) è sciolta in un’altra (solvente).
In chimica conviene fare avvenire reazioni in soluzioni poiché gli atomi, muovendosi, possono reagire con maggior facilità.
Per poter attuare una misurazione in una reazione o fare vari calcoli stechiometrici sono molto importanti i vari rapporti della concentrazione:
Per esprimere i vari rapporti è necessario utilizzare delle varie unità di concentrazione.
La più nota ed utilizzata è la molarità (M) che è data dal rapporto tra le moli di soluto e il volume di soluzione in cui esse sono contenute:
![]()
Di minore importanza, ma altrettanto utilizzata è la molalità (m), che esprime il rapporto tra le moli del soluto in una massa in kg di solvente.
![]()
la frazione molare esprime il numero delle moli di una sostanza rapportato alle moli dell’intera soluzione:
![]()
Molto utilizzata è anche la percentuale in massa, che è data dal rapporto della massa di una certa sostanza sulla massa totale della soluzione moltiplicata per 100:
![]()
Altre unità di misura sono:
Tra i vari soluti ve ne possono essere alcuni che si dissociano in ioni quando vengono posti in soluzione, conducendo elettricità: sono dette sostanze elettrolite.
Le molecole che invece non si dissociano in acqua e non conducono elettricità sono dette non elettroliti.
Talvolta è possibile che il solvente non riesca più a disciogliere il soluto e ne rimane una piccola quantità non disciolta:
La costante di equilibrio della reazione di un non elettrolita in soluzione è kc=[NE](sol).
In una soluzione con elettroliti si ha il cosiddetto prodotto di solubilità:
In generale, per i sali il prodotto di solubilità è il prodotto delle concentrazioni degli ioni, ciascuna elevata al proprio coefficiente stechiometrico.
La solubilità dipende da:
La notevole solubilità dei nitrati spiega il motivo della loro assenza nei minerali.
La scarsa solubilità dei fosfato fornisce un vantaggio allo scheletro del corpo umano, formato principalmente da fosfato di calcio, poiché gli conferisce la rigidità.
La dipendenza della solubilità dalla natura del solvente è riassunta con una regola che “il simile scioglie il simile”:
Nei gas la pressione influisce moltissimo sulla solubilità:
La legge di Henry stabilisce che la solubilità di un gas in un liquido è proporzionale alla pressione parziale del liquido stesso.
![]() ,
,
Dove S è la concentrazione molare del gas disciolto e kH è la costante di Henry, che dipende da:
Questo aumento della solubilità dovuto ala pressione si può spiegare in termini di equilibrio dinamico tra le molecole del gas e quelle della soluzione:
Per quanto riguarda l’effetto della temperatura:
un comportamento di un solido può essere definito endotermico quando ha bisogno di energia per potersi sciogliere.
Quando invece nello scioglimento viene ceduta energia, allora la reazione è esotermica.
Alcuni solidi si sciolgono endotermicamente, altri esotermicamente.
Il processo di dissoluzione in un soluto avviene in due stadi:
Una sostanza che si scioglie endotermicamente è più solubile all’aumentare della temperatura, una sostanza che si scioglie esotermicamente è meno solubile all’aumentare della temperatura.
Normalmente una dissoluzione avviene a temperatura costante:
L’entalpia di idratazione è maggiormente esotermica con:
Questa è regolata dalla legge di Born.
L’effetto di un soluto disciolto in una soluzione produce quattro cambiamenti tra loro correlati:
una proprietà colligativa è una proprietà che:
Per quanto riguarda il numero delle particelle:
per la legge di Raoult:
![]()
La causa di questo fenomeno è data dalla quantità di molecole di soluto in superficie, che diminuiscono la velocità del distacco.
L’innalzamento del punto d’ebollizione è proporzionale alla molalità (m) della soluzione
![]()
kb è la costante ebulloscopica del solvente.
Considerare la molalità in termini di ioninei composti disciolti!!
Un soluto diminuisce il punto di congelamento del solvente, determinando il cosiddetto abbassamento crioscopico.
Questo fenomeno è causato da:
Anche la temperatura di solidificazione è proporzionale alla molalità:
![]()
Il fenomeno dell’osmosi è il passaggio di solvente attraverso una membrana semipermeabile in una soluzione più concentrata.
La pressione necessaria per arrestare il flusso di solvente è detta pressione osmotica.
La causapuò essere spiegata in questi termini:
Il calcolo della pressione osmotica è dato dalla legge:
P V=nRT
in cui il numero di moli degli elettroliti deve essere duplicato in vista della dissociazione.
La cinetica chimica è lo studio delle velocità delle reazioni chimiche e dei meccanismi di reazione con cui queste avvengono.
Lo studio dei meccanismi di reazione prevede la caratterizzazione dei possibili intermedi, che comportano la formazione di prodotti intermedi.
La velocità è definita come la variazione della concentrazione dei prodotti e dei reagenti nell’unità di tempo.
La cinetica chimica si occupa di studiare la velocità con cui avviene una reazione e i fattori che la influenzano:
Le velocità di reazioni non possono essere determinate a priori, ma solamente in via sperimentale.
La velocità è quindi data dalla legge ![]() :
:
La velocità di reazione dipende dai seguenti fattori:
La stessa velocità cambia con il tempo:
La velocità di reazione in cinetica chimica si può anche esprimere con la formula:
![]()
In cui k rappresenta la costante di velocità specifica per quella reazione.
La ordine di reazione è dato dalla somma degli esponenti a cui si elevano le concentrazioni.
Si dimostra sperimentalmente che le reazioni del primo ordine hanno velocità pari a ![]() .
.
Sono di primo ordine tutte le velocità che hanno un solo reagente, così come i decadimenti radioattivi.
SI ha quindi una espressione cinetica del tipo
![]()
che separando termini da che ![]() ed integrando si ottiene:
ed integrando si ottiene:
![]() .
.
Poiché al tempo t=0 la concentrazione sarà quella iniziale [A]0, quindi
![]()
che trasformata debitamente darà la formula finale, ovvero che
![]() , che può anche essere espressa con
, che può anche essere espressa con ![]()
Ha senso quindi parlare di tempo di dimezzamento, ovvero il tempo necessario affinché la concentrazione sia la metà di quella iniziale, seguendo la formula:

Si utilizza spesso l’emivita, poiché è un modo semplice e pratico per esprimere le velocità.
La concentrazione del reagente diminuisce quindi in maniera esponenziale. Per rettificare il grafico con pendenza -k, occorrerà esprimere in funzione del logaritmo del rapporto tra la concentrazione iniziale ed una data concentrazione, che varia nel tempo costantemente.
Si ha una reazione del secondo ordine se le equazioni derivate assumono la forma:
Nel tempo, una reazione come la (1) varia secondo una legge 
1/[A] in funzione del tempo da una linea retta.
Le reazioni di ordine zero sono reazioni in cui la velocità non varia con il variare della concentrazione.
La concentrazione varia in funzione del tempo con un andamento rettilineo.
Per queste reazioni la velocità è espressa come v = k.
Affinchè delle molecole reagiscono sono necessari due procedimenti:
Siccome il numero degli urti è proporzionale alla loro concentrazione si può scrivere freq.collisioni = K[A][B],
Se tutti gli urti avessero delle collisioni efficaci la K sarebbe identica alla k della velocità di reazione, quindi la frequenza delle collisioni sarebbe identica alla velocità.
Tuttavia non sempre gli urti tra molecole risultano efficaci. Gli urti efficaci avvengono solamente se le molecole possiedono una energia superiore all’energia minima che li rende efficaci, l’energia di attivazione.
Con una distribuzione di Maxwell è possibile intuire come si trovino le molecole:
![]()
Considerando due fattori si giunge alla nota equazione di Arrhenius:
![]()
Questa legge mette parecchio in risalto il fatto che la velocità di una reazione dipenda dalla temperatura.
La velocità di una reazione chimica aumenta sempre con la temperatura, quindi, perché aumenta il numero di molecole con Ek > Ea.
Il grafico di questa equazione nella forma logaritmica ci da la possibilità di osservare la velocità di una reazione chimica in funzione della temperatura, con l’andamento di una retta:
![]()
Riportando nel grafico i valori di lnk in funzione di 1/T, si ottiene una retta che ha pendenza –Ea/R, che intercetta l’asse delle ordinate nel punto lnA.
Con un alta energia di attivazione, la pendenza aumenta, quindi la velocità aumenta velocemente all’aumentare della temperatura.
Secondo la teoria del complesso attivato, quando le molecole iniziano ad interagire, le lunghezze di legame e la disposizione geometrica cambiano e si giunge ad una configurazione della complesso attivato (o stato di transizione).
In quell’istante, si forma un complesso estremamente instabile, con un picco di energia potenziale molto più alta di quella di reagenti e prodotti.
Proprio per l’estrema instabilità, il complesso attivato tende a cambiare configurazione, assumendo quella iniziale o quella di prodotto:
Ovviamente, quindi, con una bassa energia di attivazione, si potrà formare un maggior numero di complessi attivati e la reazione procederà con una maggiore velocità.
La variazione di energia tra reagenti e prodotti non comprende l’Ea, e può essere:
Per parlare di meccanismo di reazione occorre introdurre il concetto di molecolarità:
Se la molecolarità coincide con l’ordine di reazione allora si tratta di una reazione elementare.
Talvolta si formano più stadi nel meccanismo di reazione, con la formazione di più complessi attivati:
La velocità di molte reazioni e di quasi tutte le reazioni biochimiche è aumentata dall’azione di catalizzatori, molecole proteiche ad attività catalitica.
Un catalizzatore, può accelerare solamente reazioni termodinamicamente possibili, non reazioni endoenergetiche.
Non ha alcun effetto sull’equilibrio, non ne modifica la costante, ma aumenta solamente la velocità delle reazioni.
Un catalizzatore modifica il processo di reazione, determinando un abbassamento dell’energia di attivazione, non il punto di inizio o il punto di fine.
Al termine di una reazione, il catalizzatore si trova sostanzialmente inalterato:
Occorre distinguere due tipi di catalisi:
Solamente in alcune reazioni i reagenti si trasformano completamente in prodotti:
La miscela, una volta raggiunta la condizione di equilibrio, tende a rimanere invariata nel tempo:
Data una reazione del tipo
aA + bB èç cC +dD
si chiama costante di equilibrio kc una costante specifica di una reazione, dipendente solamente dalla temperatura, data dalla legge
![]()
Data la natura matematica di questa legge, si deduce che:
Solitamente si dice che per 0,1< k <10 nessuna specie è nettamente favorita.
Per esprimere la concentrazione si possono utilizzare diverse unità di misura con differenti risultati numerici. È opportuno distinguere tra:
Se le specie chimiche coinvolte si presentano tutte in fase gassosa, talvolta può essere opportuno esprimere la costante di equilibrio in funzione delle pressioni parziali, poiché sono dipendenti solamente dalla molarità a pressione costante:
pV=nRT è p = n/V RT è p = [X] RT
Si ha quindi una variazione dell’equazione precedente ed una nuova costante kp.
![]()
Le due costanti stanno tra loro in rapporto a RT, secondo la legge
![]()
Se almeno una delle specie chimiche si trova in una fase differente dalle altre interne alla reazione, si parla di specie chimica.
Sono esempi:
La costante di equilibrio di queste reazioni è espressa in funzione della kp o della kc del gas, ma esprime anche le specie più condensate.
Con le variazioni delle condizioni di temperatura, pressione e concentrazione di una delle specie presenti nella reazione, gli equilibri si spostano secondo il principio di Le Chatelier:
Con una variazione di concentrazione di una delle specie:
Se in una situazione di equilibrio uno dei prodotti viene allontanato, l’equilibrio non viene più raggiunto e la reazione procede in una sola direzione, fino alla trasformazione totale di tutti i reagenti.
Per quanto riguarda le variazioni di pressione e volume:
Un aumento di temperatura ad un equilibrio chimico comporta assorbimento di calore:
L’acqua in minima parte si trova dissociata in H+ e OH-. Siccome gli ioni H+ non possono esistere da soli questi si trovano sotto forma di ioni idronio H3O+.
La costante di equilibrio è data da
![]()
Poiché a 25° C la concentrazione di acqua ionizzata è molto bassa, si può considerare l’acqua non dissociata come costante, con una concentrazione di 55,5 M (1000g/l / 18,011 g/mol = 55,5 mol/l ).
![]()
Questa nuova costante è definita prodotto ionico dell’acqua.
In acqua, se è maggiore la concentrazione di ioni idronio rispetto a quella degli ossidrile la soluzione è detta acido.
Se invece la concentrazione degli ossidrili è superiore a quella degli ioni idronio la soluzione è detta basica.
La neutralità si ha quando le due concentrazioni sono identiche.
Secondo Arrhenius un acido è una sostanza che in soluzione acquosa rilascia ioni H+.
Una base di Arrhenius è una sostanza che in soluzione acquosa è in grado di liberare ioni OH-.
L’ammoniaca si comporta da base perché nella reazione con acqua acquista un protone e rilascia ioni OH-:
NH3 +H2O è NH4+ + OH-
Ma la definizione di Arrhenius non spiega il perché, poiché non è l’ammoniaca a rilasciare lo ione OH-.
Si deve giungere ai concetti di acido e base coniugata, secondo la definizione di Brownsted e Lowry.
La definizione di Brownsted e Lowry amplia di parecchio il campo di applicazione della teoria di Arrehnius.
Definiscono:
Quando un acido ha perso un suo protone può però a sua volta diventare accettare, quindi base: si chiama base coniugata di un acido, quell’acido che ha perso uno o più protoni.
Si dice altresì acido coniugato una base che ha già accettato uno o più protoni ed è in grado di donarli.
Lewis definisce:
In base a questa teoria, tra un acido ed una base può formarsi un legame covalente.
Siccome la concentrazione dell’acqua rimane sostanzialmente costante si definisce una costante di dissociazione acida ed una costante di dissociazione basica:
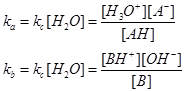
Queste due costanti possono anche essere linearizzate con i logaritmi negativi in base 10:
![]()
Queste sono in relazione con il prodotto ionico dell’acqua secondo le leggi:
![]()
Un acido è tanto più forte quanto si dissocia maggiormente. Anche una base, è più forte se si dissocia di più.
Quando è posto in soluzione acquosa, un acido:
Quando è posto in soluzione, una base:
Un acido è molto forte se la molecola presenta legami dell’idrogeno con un altro atomo con alta differenza di elettronegatività.
Un altro fattore determinante è l’energia del legame con l’idrogeno:
Per gli acidi ossigenati si individuano due categorie:
La concentrazione degli ioni H+ in soluzioni acquose assume caratteristiche molto importanti riguardo all’acidità o alla basicità della soluzione.
Piuttosto che esprimere la [H+] con numeri molto piccoli, si preferisce esprimere le potenze di 10 con logaritmi negativi in base 10, secondo la definizione
![]()
Da cui si può dedurre, secondo la legge di azione delle masse, che
![]()
Un pH può essere misurato con opportuni apparecchi detti pHmetro, che indicano la concentrazione di ioni idronio.
A livello numerico, la neutralità si ha quando la concentrazione di H+ è uguale alla concentrazione di OH-:

Se la concentrazione degli ioni H+ è alta, si ha una soluzione acida, quindi pH<7. Se la concentrazione di ioni H+ è bassa (minore di quella di OH-) si ha una soluzione basica, con pH > 7.
Se si aggiunge in acqua un acido forte (con concentrazione maggiore di 5 10-7 M), poiché questo si dissocia completamente, si può utilizzare la formula:
pH = pCacido
La stessa cosa si fa se si aggiunge una base forte:
pH = 14 –pCbase
Se invece si è in presenza di acidi o basi deboli si deve considerare anche la costante di dissociazione, poiché non li si troverà tutti dissociati.
In presenza di una soluzione acquosa con un acido debole:
pH = (pCacido+ pKa)/2
la medesima cosa si ha in presenza di una soluzione acquosa con una base debole:
pH = 14 - (pCbase+ pKb)/2
Le concentrazioni di ioni idronio o ossidrili, come formule inverse per gli acidi e le basi deboli, saranno date da:
Se si è nel caso di acidi poliprotici, ovvero quegli acidi che possono dissociare più di uno ione H+, la ka sarà data dal prodotto delle k di prima, seconda, terza…n-esima dissociazione, ed in base a quella si potranno fare i debiti calcoli.
un sale è un composto ionico che si viene a formare per reazione di un acido e una base.
I sali, poiché sono composti ionici, in acqua si dissociano completamente:
il termine idrolisi salina indica la dissociazione di molecole d’acqua indotta dagli ioni liberati da un sale.
Alcuni sali, si dissociano in ioni che non possono in alcun modo accettare o donare protoni.
Ad esempio, NaCl deriva dalla reazione di un acido e una base molto forti:
Il risultato è che il pH dell’acqua resta inalterato.
Altri sali, come ad esempio l’acetato di sodio, derivano da un acido debole e da una base forte:
In caso di sali basici si ha un aumento del pH.
Si può dimostrare che vi è un’equazione che mette in relazione la costante dell’acido debole con quella della sua base coniugata:
![]()
tramite una costante Ki detta “costante di idrolisi” che altro non è se non la costante di dissociazione basica dello ione coniugato all’acido debole.
Poiché lo ione acetato, in questo esempio, è pari agli ossidrili che si vengono a formare (trascurando quelli dell’autoprolisi dell’acqua) e un sale si dissocia completamente si può affermare che:
[CH3OO-]=Csale
in cui Csale è la concentrazione del sale disciolto.
Si avrà dunque che per la base coniugata all’acido debole:

Per i sali acidi, ovvero quelli derivanti da un acido forte e da una base debole, si assisterà invece ad una diminuzione del pH, poiché la base debole tenderà a cedere gli ioni H+.
Le equazioni rimangono
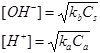
Se il sale deriva da un acido debole e da una base debole, il pH della soluzione dipende dalle costanti di dissociazione degli ioni che si liberano:
Quando sono presenti in soluzione differenti ioni appartenenti ad una specie comune, ad esempio un sale basico ed un acido debole da cui il sale basico è derivato, si utilizza la equazione di Henderson-Hasselbach, che prevede:
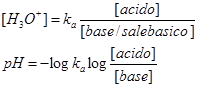
è importante notare che:
Si nota che in una soluzione quando sono presenti un acido e il suo sale (base coniugata) le cui concentrazioni differiscono per meno di un ordine di grandezza, il pH della soluzione varia molto poco variando le concentrazioni relative delle due componenti disciolte.
Se sono presenti un acido, ed il suo sale coniugato con una base forte, le variazioni di pH sono molto piccole quando si aggiungono piccole quantità di una delle due specie:
Il sangue ed altri liquidi biologici devono essere mantenuti il più possibile a pH vicino a 7,4:
Anche per calcolare l’efficacia dei tamponi si utilizza l’equazione di Hendersson-Hasselbach.
La capacità tampone è la quantità di un acido o una base che può essere aggiunta a una soluzione tampone senza fare cambiare il valore del pH oltre un valore predefinito.
Il sistema acido/base coniugata ha capacità tampone quando il rapporto delle concentrazioni (Ca/Cb) è compreso tra 0,1 e 10.
Quindi il campo di pH in cui un determinato tampone è operativo è di pK= ± 1.
Le capacità tampone sono legate alle concentrazioni assolute dell’acido e della base coniugata:
La termodinamica studia gli scambi di energia che avvengono durante una reazione chimica.
Tutte le reazioni chimiche rispettano due principi fondamentali sulla conservazione:
L’energia è definita come la capacità di un sistema di compiere un lavoro.
L’energia si trova in varie forme:
L’energia interna di un sistema è data dalla somma di tutti questi tipi di energia che questo possiede.
L’energia si misura in Joule [kg m2/s2], ma può anche essere espressa in calorie:
La legge della conservazione dell’energia assume che:
Per definire meglio gli oggetti di studio si distingue tra:
In relazione all’ambiente si distinguono tre tipi di sistemi:
Per definire un sistema termodinamico si utilizzano dei parametri detti variabili di stato: quando questi sono definiti si parla di uno stato termodinamico:
Alcune variabili di stato, per le peculiari caratteristiche sono dette funzioni di stato:
Quindi, la variazione di energia in un sistema interno è:
![]()
Volendo riferirsi ad un sistema chiuso come quello utilizzato nelle reazioni chimiche, si ha:
![]()
Ovvero che la variazione di energia di un sistema chiuso è data dalla somma del calore assorbito, e del lavoro fatto sul sistema (oppure compiuto dal sistema, con lavoro negativo).
La convenzione sui segni prevede che, per il calore:
La convenzione sui segni prevede che, per il lavoro:
Calore e lavoro ad una pressione costante
Si prenda ad esempio il lavoro meccanico fatto da un gas che si espande muovendo un pistone che si considera privo di massa. Il suo lavoro sarà dato da:
L = - pDV
Il meno è perché è lavoro compiuto dal sistema. Si evince da questa legge che il lavoro compiuto è attuato dal sistema.
Questa legge fornisce un lavoro espresso in [ l atm ], che per essere convertiti in joule devono essere moltiplicati per il fattore di conversione
1 atm = 101,3 J
Ne il lavoro né il calore scambiati dal sistema sono funzioni di stato:
Le variazioni di energia saranno date da:
![]()
Poiché la variazione di energia sarà uguale per entrambe le differenti trasformazioni si può scrivere
Q1 = Q2 + L2
Il calore ed il lavoro sono dunque proprietà del sistema che esistono solamente durante una trasformazione, quindi non sono funzioni di stato.
La variazione di energia interna, data dalla somma tra calore e lavoro, è una funzione di stato.
Entalpia e primo principio della termodinamica.
Quasi tutte le reazioni di nostro interesse avvengono a pressione costante:
Se una reazione non ha né reagenti, né prodotti, la variazione di volume può essere trascurata e considerata nulla.
Si ipotizzi di avere una reazione siffatta: reagenti è prodotti.
La variazione dell’energia interna è descritta da DE = Qp - pDV
Si può scrivere, dopo opportuni calcoli che
Ef – Ei = Qp – pVf + pVi
Qp = (Ef + pVf) - (Ei + pVi)
Quindi, l’espressione E + pV è una equazione di stato (formata da tre variabili di stato) che si definisce entalpia:
H = E + pV
Esprimibile anche come:
Hf – Hi = DH = Qp
È quindi interessante notare che la variazione di entalpia coincide con la quantità di calore scambiato, quando:
Poiché la pressione è costante, e intervengono solamente variabili di stato (dipendono solo dallo stato iniziale o finale), si può scrivere anche:
DH = DE + pDV
Utile sottolineare che se nella reazione non si presenta lo stato gassoso si può considerare nulla la variazione di volume, esprimendo la variazione di entalpia pari alla variazione di temperatura:
DH = DE
Si possono quindi definire le reazioni secondo la variazione di entalpia:
Entalpia di formazione standard
Per poter paragonare le variazioni di entalpia delle varie reazioni è necessario riferirsi a dei parametri standard di temperatura e pressione: 1 atm e 25°C.
Ogni reazione chimica è quindi caratterizzata da un proprio DH:
L’entalpia di formazione standard di un elemento è la variazione di entalpia che si ha con la formazione del composto a partire dagli elementi costituenti.
Per determinare le variazioni i entalpia standard si seguono due convenzioni:
La variazione di entalpia che si ottiene facendo reagire i due elementi è l’entalpia di formazione standard del composto.
Legge di Hess
La legge di Hess afferma che: “il calore messo in giuoco, a pressione costante, in una reazione chimica con solo lavoro di espansione, è lo stesso sia che la reazione avvenga in un solo stadio, sia che avvenga in più stadi”.
Contano dunque solamente lo stato iniziale e lo stato finale.
Energia di legame
È l’energia che si identifica con l’energia necessaria per rompere il legame stesso:
Alcune reazioni possono avvenire spontaneamente, altre no. Ciò non significa che le reazioni spontanee siano per forza esotermiche, ovvero che debbano passare ad uno stato di minore energia interna.
Per chiarire il concetto di spontaneità occorre ricorrere ad un’altra grandezza termodinamica, l‘entropia, che stabilisce il grado di disordine di un dato sistema:
Al contrario dell’energia interna e dell’entalpia, è possibile determinare il valore assoluto di entropia della sostanza, purché in condizioni standard.
Ovviamente, quando ci si discosta dalle condizioni standard il valore dell’entropia cambia:
Anche l’entropia è una funzione di stato, la cui variazione tra due punti è:
DS=Sf-Si
L’aumento di entropia (DS>0) comporta un aumento del disordine, mentre la diminuzione (DS<0) comporta il passaggio ad uno stato più ordinato.
Legge dell’aumento di entropia
La legge dell’aumento di entropia, detta anche secondo principio della termodinamica è espressa:
In altri termini:
L’entropia dell’universo tende quindi a crescere, e può essere matematicamente espressa dalla legge:
DSuniv=DSsist+DSamb
che per una reazione spontanea sarà positiva, mentre per un processo all’equilibrio sarà nulla.
Variazione di entropia in un sistema costituito da reazioni chimiche
Si consideri una reazione che rappresenta il sistema del tipo
aA è bB
La variazione di entropia che comporta è data da
DSsist= DSreaz= bS0(B) – aS0(A)
ovvero la differenza tra le entropie standard dei prodotti e dei reagenti, moltiplicate per il coefficiente stechiometrico.
In sunto si possono trarre le seguenti considerazioni generali:
Variazione di entropia dell’ambiente
Si considera la relazione esistente tra entropia e entalpia.
Quando in un sistema avviene un processo esotermico:
Se in un sistema isolato avviene un processo endotermico:
Per i processi che avvengono a pressione costante, la variazione di entalpia è uguale al calore scambiato, quindi, in virtù dell’aumento o della diminuzione del disordine, l’entropia e l’entalpia sono legate dalla legge:
![]()
Se si suppone che sia l’ambiente che il sistema siano alla medesima temperatura T.
Con i tre metodi operativi imparati si può trovare la variazione di entropia dell’universo secondo la legge:
DSuniv=DSsist+DSamb
I tre metodi sono:
Il terzo principio della termodinamica afferma che l’entropia di una sostanza cristallina pura allo zero assoluto è nulla.
S0 K= 0
Si è visto che:
Tuttavia, si possono considerare altri movimenti delle molecole che non sono solo presenti allo stato gassoso:
Questi moto sono un modo delle molecole di accumulare energia:
Ludwig Boltzman propose una formula che tiene conto di queste considerazioni, i cui termini sono:
S = K ln W
Per grado di movimento 1, ovvero per cristalli puri che non presentano alcun movimento, l’entropia, secondo questa legge, è nulla.
Il terzo principio della termodinamica permette di calcolare l’entropia assoluta di una sostanza, poiché:
![]()
Dove Sf è proprio l’entropia assoluta.
Funzione di Gibbs: l’energia libera.
Per compiere un calcolo riguardo alla variazione di entropia dell’universo non è strettamente necessario operare facendo calcoli relativi all’ambiente e al sistema.
È possibile riferirsi solamente al sistema, trovando determinate equazioni. Per processi spontanei si sa che:
DSuniv=DSsist+DSamb > 0
e sostituendo al DSambil termine ![]() si ottiene
si ottiene
![]()
che è espresso solamente con funzioni relative al sistema. Cambiando di segno e operando opportuni raccoglimenti si ottiene:
![]()
Poiché i termini ![]() , si può definire una nuova funzione di stato, detta energia libera di Gibbs, indicata con G, la cui variazione, a temperatura costante, è
, si può definire una nuova funzione di stato, detta energia libera di Gibbs, indicata con G, la cui variazione, a temperatura costante, è
![]()
che può anche essere espressa nella forma
![]()
Come si può vedere, il calore prodotto in un processo (la variazione di entalpia) è utilizzabile in maniera differente:
Non tutto il calore prodotto in un sistema può essere utilizzato per compiere lavoro.
Energia libera e spontaneità di una reazione.
In funzione della variazione di energia libera possiamo indicare la spontaneità di una reazione:
Avendo la formula dell’energia libera, si possono esaminare in base a questo schema quando la reazione o il processo è spontaneo. Si facciano delle disequazioni con la legge:
![]()
Energia libera e costante di equilibrio.
L’energia libera è una grandezza estensiva, dipende cioè dalla quantità di materia presa in considerazione.
Per facilitare il confronto tra le varie sostanze ci si riferisce ad una energia libera molare standard, ovvero a grandezze espresse con la lettera G0, di:
In maniera analoga, per una reazione si esprime la variazione di energia libera standard con il termine DG0, definita come:
![]()
Sempre se la reazione avvenga in condizioni standard (25°C e 1 atm per una mole di reagente).
Per calcolare il DG di una reazione occorre conoscere:
Per una sostanza singola, G è espressa dall’equazione
Gx = G0 + RT ln[X]
Per una reazione, la variazione di energia libera, sarà quindi data dalla legge:
![]()
Il segno della variazione di energia libera di una reazione non dipende solamente dalla variazione standard, ma anche dal segno del logaritmo naturale del rapporto tra le concentrazioni dei prodotti e dei reagenti.
Poiché all’equilibrio DG=0, si ha che
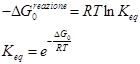
equazione di van’t Hoff
Dall’equazione precedente, deriva un’equazione che, noti i valori di due costanti di equilibrio a date temperature, è possibile risalire alla variazione di entalpia:
![]()
Energia libera: considerazioni generali e reazioni accoppiate.
Si può dimostrare che alcune reazioni con una variazione standard di energia libera positiva, possono essere spontanee se inserite in un ciclo di reazioni, dette reazioni accoppiate.
Dal punto di vista biochimico, questo tipo di reazioni è molto interessante:
In una reazione red-ox, è bene notare che:
Quando un elemento si ossida, perde due elettroni. Dopo essersi ossidato può potenzialmente ricevere nuovamente i due elettroni persi, quindi ridursi. La medesima cosa avviene con un elemento che si riduce: acquista elettroni ed in seguito è potenzialmente disposto a cederli.
Questo concetto permette di parlare di coppie coniugate di ossidoriduzione:
Quel metodo numerico esiste ed ha le basi nell’elettrochimica, e permette di:
Affinché la materia possa condurre corrente elettrica devono essere soddisfatte due condizioni:
Nei metalli sono gli elettroni liberi dal reticolo cristallino che si muovono sotto l’azione di una ddp.
Nelle soluzioni sono gli ioni disciolti che si muovono nel liquido sotto l’azione di una differenza di potenziale.
Il processo di elettrolisi permette il trasporto di carica in soluzione e avviene quando vi sono ioni in una soluzione:
Nell’elettrolisi del cloruro di sodio (NaCl) avvengono queste due semireazioni:
Na+ + e è Na
2Cl- è Cl2 + 2e
è necessario aggiungere che affinché si verifichi il transito degli ioni è necessario raggiungere una differenza di potenziale minina, detto potenziale di scarica, che è caratteristico dello ione in determinate condizioni sperimentali.
Se sono presenti sia ioni negativi che positivi, la scarica si dirige in maniera preferenziale verso lo ione che richiede minor energia.
La carica di un elettrone è pari a 1,602 10-19 coulomb, e per ridurre un grammoatomo di qualsiasi ione positivo, devono passare per l’anodo 1 mole di elettroni:
Per il passaggio di 1 F attraverso l’anodo dovranno passare:
Questo vale ovviamente anche per gli anioni
Si definisce equivalente elettrochimico il peso in grammi di sostanza trasformata all’elettrodo per il passaggio di un faraday.
L’equivalente elettrochimico è uguale al peso molecolare o atomico diviso per il numero di elettroni scambiati durante la trasformazione.
Queste considerazioni portano alle due leggi di Faraday:
Le due leggi di Faraday possono essere riunite in:
La reazione tra Zn metallico e solfato di rame produce:
Le due semireazioni possono essere fatte avvenire in ambienti separati:
In tal modo si può ottenere la trasformazione di energia chimica in energia elettrica.
L’apparato che permette la produzione di energia per mezzo di una redox spontanea è detto cella elettrolitica o pila.
Per convenzione gli elettrodi vengono chiamati:
Affinché non si verifichi la saturazione di ioni positivi in una cella (Zn2+) e di ioni negativi nell’altra (SO4-), si inserisce tra le due celle un tubo ad U detto ponte salino, che contiene un sale che rilascia ioni per bilanciare le cariche.
Avviene dunque:
Il fatto che gli elettroni fluiscano da una cella all’altra indica la presenza di una ddp che ne permette il flusso tra gli elettrodi:
Il modo convenzionale per rappresentare una cella è:
Zn(s) / Zn2+(sol) // Cu2+(sol) / Cu(s)
Le linee diagonali semplici indicano le separazioni di fase, quelle doppie il ponte salino.
L’energia elettrica che passa per una cella è data dal prodotto della ddp per la carica che la attraversa:
energia elettrica = DE q
in cui DE è la differenza di potenziale, espressa in volt e q la carica in coulomb che può anche essere data da q=nF, quindi
energia elettrica = DE n F
In un processo spontaneo la variazione di energia libera è negativa, e per la reazione precedente può essere espressa da:
-DG = DE n F
![]()
Dopo opportuni calcoli, eguagliando le due precedenti si ottiene l’equazione di Nerst, che è espressa come:
![]()
in cui:
I potenziali di elettrodo vengono per convenzione sempre espressi come potenziali di riduzione standard:
I potenziali di riduzione ordinano le sostanze in base al loro potere riducente o ossidante:
Una serie elettrochimica è in ordine decrescente di potere ossidante e crescente di potere riducente.
elettrodi metallici.
Gli elettrodi a rame e zinco fino a ora analizzati sono elettrodi metallici, costituiti da metalli immersi in soluzioni con i propri ioni.
Per avere i potenziali di un elettrodo con una reazione metallica ione/metallo, si utilizza l’equazione di Nerst.
![]()
inoltre, a 25°C si po’ trasformare l’equazione precedente in
![]()
elettrodi a gas.
Tipici elettrodi di questo tipo sono gli elettrodi ad idrogeno, fluoro e cloro, basati su reazioni del tipo:
2H+(sol) + 2e-èç H2(g)
In questo tipo di elettrodi, la specie gassosa viene fatta gorgogliare a 1 atm intorno ad una lamina di platino immersa nella soluzione contenente la stessa specie in forma ossidata (nel caso dell’H+) o ridotta.
La lamina di platino ha la funzione di assicurare una superficie su cui avvenga la reazione che può consentire il contatto esterno.
L’equazione di Nerst, ad esempio per l’H è applicata così:
![]()
![]()
Elettrodi di materiale inerte immersi in una soluzione con semicoppia ossidoriduttiva
Una lamina di platino (Pt) è immersa in una soluzione contenente sia ioni ferrosi che ioni ferrici.
La superficie di platino immersa in una soluzione che contiene i due ioni fa da tramite tra le due specie e assume un potenziale in funzione delle loro concentrazioni relative:
![]()
in cui lo ione ferroso è ‘elemento ridotto e lo ione ferrino è quello ossidato.
La ddp che determina il passaggio di elettroni tra due semicelle esprime la tendenza a ridursi/ossidarsi delle coppie ossidoriduttive.
Non essendo possibile misurare il potenziale di una singola semicella, si misurano le ddp degli elettrodi di una pila chimica.
Come elettrodo di riferimento è stato scelto l’idrogeno, a cui è stato posto potenziale E=0 V, in condizioni standard:
Se dunque si usa una semicella ad idrogeno e un’altra semicella con un altro elemento, la fem della cella sarà data dal potenziale dell’altro elemento.
Per convenzione il potenziale è:
In definitiva, si definisce come potenziale di riduzione standard di una certa redox la fem che si stabilisce in condizioni standard tra le semicelle dell’elemento e dell’idrogeno (campione).
Le varie coppie ossidoriduttive possono essere quindi disposte in una serie dei potenziali standard di riduzione:
I potenziali che sono positivi tenderanno ad ossidarsi (alto potere ossidante), quelli negativi a ridursi (alto potere riducente) rispetto all’idrogeno.
Una conseguenza importante è data dalla possibilità di prevedere il verso della reazione:
Per una reazione di ossidoriduzione vale la relazione
-DG = DE n F
in condizioni standard è anche vero che
![]()
Che eguagliandole permette di ottenere la relazione:
nF DE0 = RT ln Keq.
Da questa equazione si deduce che:
E’ possibile, data la legge di Nerst, costruire una pila in cui le due semicelle possono essere costituite dalla stessa coppia i reazione, ma con concentrazioni differenti.
In una semicella si ha Cu / [Cu2+]= C1, nell’altra [Cu2+]=C2/Cu.
DE = RT/nF ln(C2/C1)
Quando le due soluzioni saranno giunte ad equilibrio, l’energia elettrica cessa di circuitare.
Una pila a concentrazione di ioni idrogeno, permette grazie a questo principio, nota una concentrazione degli ioni H+ di una delle due semicelle, di determinare l’altra concentrazione di ioni H+.
La formula per calcolarla è
DE = 0,059 (pH1-pH2).
Noto uno dei due valori di pH si può giungere all’altro misurando la fem.
Fonte: http://www.bluejayway.it/Enrico_Colombos_Page/Medicina_files/CORSO%20DI%20CHIMICA.doc
Sito web da visitare: http://www.bluejayway.it
Autore del testo: Prof. Arosio
Il testo è di proprietà dei rispettivi autori che ringraziamo per l'opportunità che ci danno di far conoscere gratuitamente i loro testi per finalità illustrative e didattiche. Se siete gli autori del testo e siete interessati a richiedere la rimozione del testo o l'inserimento di altre informazioni inviateci un e-mail dopo le opportune verifiche soddisferemo la vostra richiesta nel più breve tempo possibile.
I riassunti , gli appunti i testi contenuti nel nostro sito sono messi a disposizione gratuitamente con finalità illustrative didattiche, scientifiche, a carattere sociale, civile e culturale a tutti i possibili interessati secondo il concetto del fair use e con l' obiettivo del rispetto della direttiva europea 2001/29/CE e dell' art. 70 della legge 633/1941 sul diritto d'autore
Le informazioni di medicina e salute contenute nel sito sono di natura generale ed a scopo puramente divulgativo e per questo motivo non possono sostituire in alcun caso il consiglio di un medico (ovvero un soggetto abilitato legalmente alla professione).
"Ciò che sappiamo è una goccia, ciò che ignoriamo un oceano!" Isaac Newton. Essendo impossibile tenere a mente l'enorme quantità di informazioni, l'importante è sapere dove ritrovare l'informazione quando questa serve. U. Eco
www.riassuntini.com dove ritrovare l'informazione quando questa serve