I riassunti , gli appunti i testi contenuti nel nostro sito sono messi a disposizione gratuitamente con finalità illustrative didattiche, scientifiche, a carattere sociale, civile e culturale a tutti i possibili interessati secondo il concetto del fair use e con l' obiettivo del rispetto della direttiva europea 2001/29/CE e dell' art. 70 della legge 633/1941 sul diritto d'autore
Le informazioni di medicina e salute contenute nel sito sono di natura generale ed a scopo puramente divulgativo e per questo motivo non possono sostituire in alcun caso il consiglio di un medico (ovvero un soggetto abilitato legalmente alla professione).
SPETTROSCOPIA
I vari tipi di spettroscopia utilizzano le interazioni tra radiazioni elettromagnetiche e materia per ottenere dettagliate informazioni analitiche, strutturali ed elettroniche del materiale in esame. La materia infatti può assorbire, emettere, diffondere, diffrangere le radiazioni in base alle proprie caratteristiche chimiche e fisiche. Le più comuni tecniche spettroscopiche (IR, UV, NMR, spettri atomici) sfruttano il fenomeno dell’assorbimento da parte della materia di specifiche radiazioni. Tuttavia anche gli altri tipi di interazioni possono essere utilizzati vantaggiosamente (diffrazione dei raggi X, spettroscopia Raman, Fluorimetria e spettri atomici di emissione, etc.).
Affinché la materia assorba radiazione elettromagnetica (e la relativa energia), è necessario che si attivino determinate transizioni la cui natura dipende proprio dall’energia del fotone incidente (correlabile a frequenza, numeri d’onda, e lunghezza d’onda).
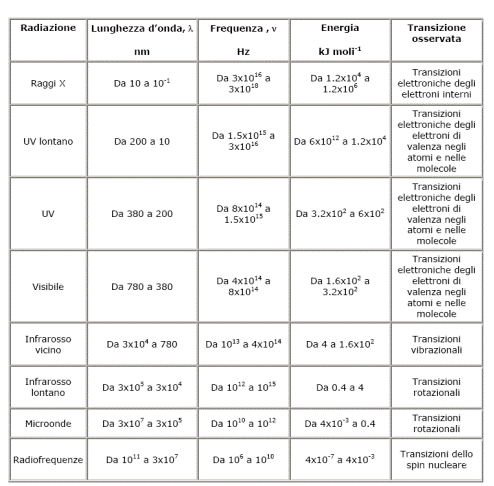
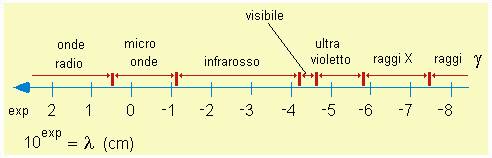
Le due più comuni tecniche spettroscopiche studiano l’assorbimento di radiazioni IR (spettroscopia vibro-rotazionale) ed UV (spettroscopia elettronica).
SPETTROSCOPIA IR
La spettroscopia IR utilizza l’interazione della radiazione elettromagnetica con i livelli vibrazionali e rotazionali (vibro-rotazionali) di sistemi molecolari. Per capire come il moto rotazionale o vibrazionale possa fornire informazioni sulla struttura delle molecole; si deve esaminare il problema mediante un modello fisico che lo rappresenti:

Tale modello assimila la molecola biatomica ad un oscillatore armonico, cioè ad un sistema costituito da due masse m1 m2 tenute insieme da una molla elastica (il legame chimico). Tale sistema si troverà in una condizione di equilibrio, nella quale la molla mantiene ad una certa distanza x° le due sfere (x°=distanza di equilibrio).
Per vibrazioni armoniche, la forza di richiamo elastica è descrivibile dalla LEGGE DI HOOKE:
F = -KDx
Dove Dx è la variazione della distanza tra i due atomi nel sistema rispetto a quella d’equilibrio; K è la costante di forza. L’equazione è scritta con il segno meno per mostrare esplicitamente che, sebbene F agisca nella direzione opposta allo spostamento subito dalla molla, la costante di proporzionalità K è positiva. La forza di richiamo funziona sia in estensione che in compressione. Il verso cambia, ma il modulo è sempre lo stesso. Assimilare K alla forza di legame è improprio poiché il legame più che avere una forza va valutato in termini di energia (energia guadagnata per la formazione del legame o energia necessaria per rompere un legame). Le due masse sotto l’azione di questa molla si muovono con un moto oscillatorio periodico. Per ottenere un’equazione che definisca la frequenza di questo oscillatore si deve rappresentare la molecola biatomica come un’ unica entità fisica la cui massa corrisponde alla cosiddetta massa ridotta del sistema:
m=m1 m2 da cui:
m1+m2
n1=n2=nclass=1/2p(K/m)1/2
Tale equazione mostra che la frequenza di vibrazione è direttamente proporzionale alla costante di forza K e inversamente proporzionale alla massa ridotta m. Di conseguenza un sistema costituito da due atomi vibrerà con una frequenza nclass tanto più elevata quanto più forte sarà il legame e quanto più piccola sarà la massa ridotta del sistema. Tale equazione, risulta utile per calcolare la frequenza di vibrazione di una coppia di atomi legati.
L’energia totale di questo oscillatore armonico sarà data dalla somma dell’energia cinetica e dell’energia potenziale. Secondo la legge di Hooke, l’energia potenziale è data dalla seguente espressione:
F= - dU
Dx
U=-ò F d(Dx) = ò K(Dx) d(Dx) (1)
Da cui, risolvendo l’integrale si ha:
U= 1/2 K(Dx)2 (2)
Se si riporta in diagramma l’energia potenziale in funzione dell’elongazione Dx, dalla posizione di equilibrio x0, si ottiene una parabola.
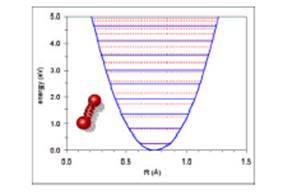
all’interno di questa parabola, i segmenti orizzontali definiscono vari livelli energetici vibrazionali. Nelle molecole questi livelli sono quantizzati. Il passaggio dall’uno all’altro deve ottemperare la legge di Plank: E=hn
TRATTAZIONE QUANTISTICA
Per un vibratore armonico microscopico l’energia è data dall’espressione:
Evibr=h nclass (v+1/2)
Dove Evibr è l’energia del livello quantico vibrazionale definito dal numero quantico v = 0,1,2,3..
Ad esempio l’energia associata allo stato v=0 cioè dello stato fondamentale, sarà :
(v= 0)+1/2=1/2 ; Evibr=1/2hnclass e cosi via.
Sostituendo a nclass= 1/2p(K/m)1/2 si ha: Evibr=h/2p(K/m)1/2(v+1/2)
Se si considera il salto da un livello energetico a quello successivo con Dv=1 (salto di variazione) l’energia che separa questi due livelli sarà:
DEvib= hnclass = h/2p*(K/m)1/2
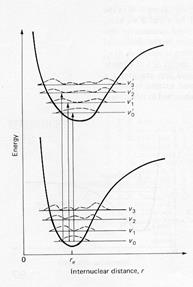
La radiazione elettromagnetica deve avere energia pari a DEvib per determinare transizioni di livelli vibrazionali, ossia per promuovere il passaggio dal livello vibrazionale fondamentale al primo eccitato. Se, infatti, da un punto di vista classico l’energia varia in modo continuo, nei sistemi atomici, i livelli energetici sono quantizzati. La condizione di equilibrio corrispondente a v=0 (livello energetico più basso) non ha energia potenziale = 0, ma 1/2 hnclass. Al crescere dell’elongazione, l’energia della molecola biatomica non può aumentare all’infinito; infatti ad un certo punto il legame si rompe ed il sistema raggiunge una condizione di energia potenziale corrispondente alla somma delle energie potenziali dei due atomi separati a distanza infinita. Tale energia è “finita” e calcolabile. Raffinando il modello, si può elaborare una curva più realistica di carattere quantitativo, che riporta sulle ascisse deformazioni in unità di Angstrom, e sulle ordinate l’energia potenziale (vedi figura). Tale curva è detta Curva di Morse. Nel diagramma è riportata la curva di energia potenziale per la molecola di HCl.
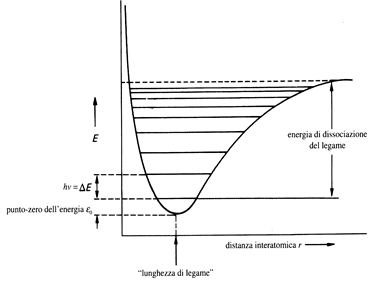

La curva di Morse prende il nome da un chimico-fisico, che per primo propose un’espressione matematica per rappresentare meglio la situazione d’ energia potenziale di un oscillatore armonico reale. Dal grafico si può notare che nel caso della:
Estensione dopo 3 o 4 Angstrom la curva si appiattisce , raggiunge un plateau (zona piatta quasi parallela all’asse delle ascisse, che corrisponde ai due atomi separati in quanto l’interazione reciproca tra gli atomi è quasi trascurabile. L’energia aumenta tanto più lentamente, quanto maggiore diventa la lunghezza del legame. La parte destra della curva di Morse, quindi risulta notevolmente differente dal modello dell’oscillatore ideale.
Compressione: La parte sinistra della curva di Morse è più vicina all’approssimazione parabolica, è solo un po’ più ripida a causa della repulsione sempre più spinta.
Un’importante differenza tra le due curve è che la differenza tra i vari livelli vibrazionali è costante nella curva di Hooke, mentre va a diminuire progressivamente in quella di Morse.
SPETTRI ROTO-VIBRAZIONALI DELLE MOLECOLE BIATOMICHE:
A temperatura ambiente (T=25°C) le molecole hanno energia sufficiente per popolare vari livelli rotazionali. Ad ogni livello vibrazionale risultano associati vari livelli rotazionali. Dal punto di vista spettroscopico questo significa che la medesima transizione vibrazionale coinvolge svariati livelli rotazionali che complicheranno lo spettro le cui bande risulteranno composte da molti picchi tra loro vicini (struttura fine rotazionale). Il fenomeno è osservato solo per campioni allo stato gassoso. In soluzione o allo stato solido i tempi per effettuare una completa rotazione sono nettamente superiori ai tempi di una transizione vibrazionale, per cui gli spettri perdono la struttura fine.
Per interpretare ed identificare una qualsiasi molecola organica sono necessarie due considerazioni:
a)Regole di selezione
b)Approssimazione di gruppo.
REGOLE DI SELEZIONE
Le regole di selezione impongono limiti restrittivi alla possibilità di interazione tra radiazione e materia. Ogni tipo di spettroscopia ha le proprie regole di selezione. Regole di selezione sono anche il principio della massima molteplicità di Hund e le regole che governano il riempimento progressivo degli orbitali.
PRIMA REGOLA DI SELEZIONE:
La transizione può avvenire solo se comporta una variazione del numero quantico v pari a Dv= ±1
Per conoscere la frequenza della radiazione che determina la transizione bisogna considerare anche il salto Dv.
n=[1/2p (k/m)1/2]Dv
Le bande proibite (poco intense) possono essere a volte riconosciute in quanto cadono a multipli delle bande permesse. Quando possono essere osservate sono di grande aiuto per l’ attribuzione della frequenza fondamentale.
SECONDA REGOLA DI SELEZIONE:
Tale regola ha a che vedere con il momento dipolare di un legame. Tale momento è il prodotto della separazione di carica q per la distanza di separazione d:
M=q*d
l’assorbimento di un oscillatore molecolare è proporzionale alla variazione di momento dipolare durante la vibrazione.
Il campo elettrico del dipolo oscilla alla frequenza di vibrazione del legame
n=1/2p(k/m)1/2
quindi consente alla molecola di interagire con il campo elettrico della radiazione oscillante alla medesima frequenza.
APPROSSIMAZIONE DI GRUPPO:
Una molecola di N atomi, comporta l'esistenza di N/2 coppie. A questo punto possiamo chiederci: è possibile considerare ogni coppia come un oscillatore armonico indipendente, oppure la molecola si comporta come un unico complesso sistema che coinvolge nelle oscillazioni tutti gli atomi? In realtà sono vere entrambe le ipotesi.
Il PRINCIPIO DELL'APPROSSIMAZIONE DI GRUPPO afferma che un oscillatore costituito da pochi atomi risulta indipendente dal resto della molecola quando la frequenza di oscillazione è molto diversa da quella degli oscillatori atomici adiacenti. Nel caso contrario l'oscillatore farà necessariamente parte di un sistema più grande, dunque l’approssimazione di gruppo non sarà più valida. L’approssimazione di gruppo è attuabile nel caso di coppie di atomi quali C-O, O-H e C-H presenti nei composti organici, i quali risultano facilmente identificabili in base alle loro specifiche masse ridotte ed energie di legame. A livello dei legami C-C non è più possibile ritenere l'approssimazione valida in quanto è difficile distinguere fra i numerosi sistemi C-C che compongono lo scheletro molecolare. In realtà esisteranno anche delle piccole bande dovute a sistemi costituiti esclusivamente da singole coppie C-C, ma sicuramente compariranno delle VIBRAZIONI DI SCHELETRO che si riferiscono al movimento e alle deformazioni dell'intera impalcatura molecolare. Tali vibrazioni costituiranno diversi picchi relativi e tutte le possibili deformazioni che esistono per una molecola nella zona chiamata ZONA DEL FINGER-PRINT (impronta digitale). Questi picchi di intensità e numero variabili risultano essere specifici per ogni singolo composto se presi nel loro insieme, rappresentando quindi un modo per identificare UNIVOCAMENTE un composto.
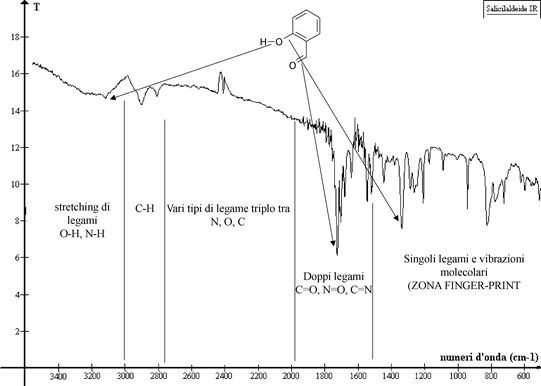
A seconda della forma di vibrazione si distingue fra:
Esempi di vibrazioni di deformazioni più complesse sono:

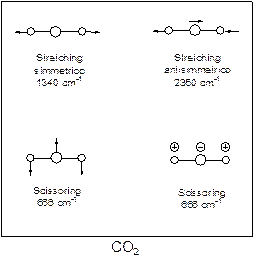 Wagging
Wagging
Spettroscopia nell’UV/ visibile
A frequenze più alte dell’IR ci avviciniamo al visibile. L’IR vibrazionale corrisponde a lunghezze d’onda di un paio di mm; la regione del visibile, invece, si estende da 800-400 nm. Ad 800 nm corrisponde la l della radiazione meno energetica (rosso), mentre 400 nm quella più energetica (blu-violetto). Subito dopo il violetto c’è l’UV, una regione estesa da 400 e 150 nm, della quale la spettroscopia tradizionale utilizza solo una piccola banda. I raggi UV sono comunemente divisi in base ai valori decrescenti di l in UVA, UVB e UVC. Questi ultimi sono estremamente pericolosi per gli esseri viventi in virtù del potere energetico dei singoli fotoni sono sufficienti a determinare la rottura di legami chimici di molecole biologicamente rilevanti. La spettroscopia UV (al contrario di quella IR) usa fotoni sufficientemente energetici da eccitare transizioni elettroniche, che provocano il passaggio degli elettroni molecolari più esterni in orbitali di antilegame.
In una molecola una transizione elettronica si accompagna sempre a transizioni di energia rotazionale e vibrazionale, perché la molecola, mentre è eccitata elettronicamente, continua a ruotare e vibrare (vedi figura). Di conseguenza una transizione elettronica molecolare non dà luogo ad una singola riga, ma ad un sistema di righe detto banda, il cui massimo di assorbimento rappresenta la lmax.
Il principio dell’UV è quello di sfruttare una radiazione per eccitare elettroni che si trovano in vari livelli energetici “fondamentali”. Al livello atomico questa spettroscopia consente di individuare alcuni metalli e questo può essere importante per verificare nell’acqua la presenza di mercurio, piombo o nell’aria la presenza di piombo tetraetile usato nelle benzine. Nella spettroscopia molecolare, consideriamo transizioni elettroniche tra livelli energetici di orbitali molecolari.
Orbitali molecolari
Sono combinazioni degli orbitali atomici la cui simmetria dipende dagli orbitali originari, ma anche dal tipo di combinazione. Gli orbitali molecolari sono di legame s e p (energia minore), di non legame n (energia intermedia) o di antilegame s* e p*(energia superiore).
Gli orbitali di non legame n sono spesso localizzati su singoli atomi.
La spettroscopia UV si occupa delle transizioni da “livelli fondamentali (popolati) a livelli eccitati (non popolati) degli orbitali molecolari” (Principalmente i salti riguardano il più alto stato occupato [HOMO] ed i più basso stato non occupato [LUMO]).
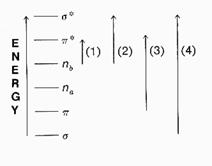
Quando una sostanza assorbe nel visibile è colorata, perché possiede uno stato eccitato separato da quello fondamentale da un DE relativamente basso (corrispondente a l tra 400 e 800 nm). Il DE risulta maggiore per transizioni che assorbono UV la cui l è minore di 400 nm.
Regole di selezione per l’UV
La spettroscopia UV presenta 3 regole di selezione, la prima è molto vicina concettualmente a quella dell’IR.
La I° regola di selezione afferma che in una transizione elettronica si deve avere una forte ridistribuzione della carica elettrica e quindi una concreta variazione del momento dipolare.
Nel caso delle transizioni elettroniche la distribuzione delle cariche è legata agli elettroni che occupano determinati orbitali, mentre i nuclei possono essere considerati fermi. Questi orbitali avranno delle geometrie diverse di densità elettronica o meglio delle y². Il passaggio di un elettrone da un orbitale molecolare all’altro influenza la distribuzione delle cariche. In realtà questa regola di selezione governa solo la probabilità della transizione (intensità della banda) visto che nessun orbitale possiede esattamente la stessa distribuzione di carica di un altro.
La II° regola di selezione ha a che vedere con lo “spin” elettronico. Noi sappiamo infatti, che l’elettrone è una particella piccola dotata di carica e di spin. Lo spin può essere di due tipi: ±1/2 e viene definito, in senso classico come la rotazione dell’elettrone intorno al proprio asse. La somma degli spin di tutti gli elettroni è definito come S cioè spin totale. Per la seconda regola di selezione lo spin totale in una transizione si deve conservare. DS = 0.
Questo significa che ci possono essere dei salti elettronici tra orbitali, ma questi non devono alterare il momento orbitalico di spin (MOS) totale.
Cioè, se ho un sistema di elettroni con spin appaiati (MOS = 0) nella transizione elettronica deve succedere qualcosa che comporti comunque un sistema che rimanga appaiato. Se ho un sistema spaiato tale si dovrà conservare.
La III° regola di selezione è detta regola della simmetria orbitalica.
Essa afferma che una transizione è permessa solamente se la simmetria della distribuzione elettronica della y² si conserva durante la transizione.
Questa regola, in parole povere, ci dice che la transizione è tanto più probabile che avvenga quanto più la simmetria tra gli orbitali è conservata.
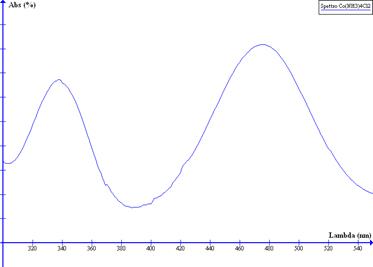
Analisi quantitative dell’UV. La legge di Lambert-Beer
L’UV trova vantaggiose applicazioni nell’ analisi quantitativa. Esiste infatti un’equazione che permette di correlare concentrazione di una sostanza in soluzione con l’ assorbanza A. L’Assorbanza è il parametro di lettura mediante il quale gli spettrofotometri visibile-UV definiscono la quantità di radiazione assorbita nel passaggio attraverso una soluzione (legge di Lambert-Beer):
dove I°= radiazione incidente, I= radiazione emergente, e = coefficiente di estinzione molare ( è molecola-specifico, indica quanto assorbe la molecola di una sostanza a c = 1M, ad una determinata l ), l = lunghezza, in cm, della cella che contiene la soluzione da esaminare, essa è detta anche “cammino ottico” della radiazione; c = concentrazione molare. Quest’ultima è l’unica incognita nella analisi quantitativa, che permette di definire la concentrazione della soluzione in base alle letture di Assorbanza .
Fonte: http://moodle2.unime.it/pluginfile.php/3695795/mod_resource/content/1/intro_spettroscopia.doc
Sito web da visitare: http://moodle2.unime.it
Autore del testo: non indicato nel documento di origine
Il testo è di proprietà dei rispettivi autori che ringraziamo per l'opportunità che ci danno di far conoscere gratuitamente i loro testi per finalità illustrative e didattiche. Se siete gli autori del testo e siete interessati a richiedere la rimozione del testo o l'inserimento di altre informazioni inviateci un e-mail dopo le opportune verifiche soddisferemo la vostra richiesta nel più breve tempo possibile.
I riassunti , gli appunti i testi contenuti nel nostro sito sono messi a disposizione gratuitamente con finalità illustrative didattiche, scientifiche, a carattere sociale, civile e culturale a tutti i possibili interessati secondo il concetto del fair use e con l' obiettivo del rispetto della direttiva europea 2001/29/CE e dell' art. 70 della legge 633/1941 sul diritto d'autore
Le informazioni di medicina e salute contenute nel sito sono di natura generale ed a scopo puramente divulgativo e per questo motivo non possono sostituire in alcun caso il consiglio di un medico (ovvero un soggetto abilitato legalmente alla professione).
"Ciò che sappiamo è una goccia, ciò che ignoriamo un oceano!" Isaac Newton. Essendo impossibile tenere a mente l'enorme quantità di informazioni, l'importante è sapere dove ritrovare l'informazione quando questa serve. U. Eco
www.riassuntini.com dove ritrovare l'informazione quando questa serve