I riassunti , gli appunti i testi contenuti nel nostro sito sono messi a disposizione gratuitamente con finalità illustrative didattiche, scientifiche, a carattere sociale, civile e culturale a tutti i possibili interessati secondo il concetto del fair use e con l' obiettivo del rispetto della direttiva europea 2001/29/CE e dell' art. 70 della legge 633/1941 sul diritto d'autore
Le informazioni di medicina e salute contenute nel sito sono di natura generale ed a scopo puramente divulgativo e per questo motivo non possono sostituire in alcun caso il consiglio di un medico (ovvero un soggetto abilitato legalmente alla professione).
GRUPPO DI LAVORO DI GENETICA CLINICA - SIGU
LINEE GUIDA PER L’INQUADRAMENTO DIAGNOSTICO DELLE IPOACUSIE GENETICHE
Ottobre 2004
Documento discusso dal Gruppo di lavoro di Genetica Clinica e preparato da:
Paolo Gasparini (coordinatore)(Genetica Medica, Seconda Università degli Studi di Napoli e TIGEM Telethon Institute of Genetics and Medicine),
Rosario Casalone (U.O.di Genetica Medica, Azienda Ospedaliera Universitaria Ospedale di Circolo e Fondazione Macchi, Varese),
Faustina Lalatta (Genetica Clinica , Istituti Clinici di Perfezionamento, Milano),
Elio Marciano (Divisione di Audiologia/ORL, Università Federico II, Napoli),
Alessandro Martini (Divisione di Audiologia/ORL, Università di Ferrara),
Maria Cristina Patrosso (Laboratorio di Genetica Medica, Ospedale Niguarda, Milano),
Lucia Perroni (Laboratorio di Genetica Umana, Ospedali Galliera, Genova),
Le ipoacusie interessano circa il 4% della popolazione di età inferiore ai 45 anni e comprendono un vasto spettro di manifestazioni cliniche. Nei paesi sviluppati, almeno il 60-70% dei casi di ipoacusia è dovuto a cause genetiche mentre la rimanente parte è dovuta a cause di natura ambientale quali infezioni durante la gravidanza, traumi, farmaci, etc. come riportato qui di seguito:
In epoca prenatale: Citomegalovirus, Rosolia, Farmaci ototossici
In epoca neonatale: Meningite, Prematurità, Ipossia, Ittero neonatale
In epoca postnatale: Neoplasie, Infezioni,Traumi, farmaci ototossici, patologie vascolari
Oltre il 60% delle sordità è di origine genetica, di queste, il 30% fanno parte di una condizione sindromica, ad ereditarietà mendeliana o mitocondriale, quali ad esempio:
Sindrome di Alport
Sindrome di Norrie
Sindrome di Usher
Sindrome di Pendred
Sindrome di Waardenburg
Sindrome di BOR
Sindrome di Jervell-Lange-Nielsen
Il 70% sono forme non sindromiche ad ereditarietà mendeliana o mitocondriale suddivise come di seguito
Recessive (circa l’80% dei casi)
Dominanti (circa il 20% dei casi)
X-linked (circa l’1% dei casi)
Mitocondriali (circa l’1% dei casi)
I genetisti possono contribuire all’identificazione dell’eziologia e alla definizione del rischio genetico dei pazienti affetti da ipoacusie genetiche e dei loro familiari, facilitando la realizzazione di un percorso integrato con altri specialisti orientato alla diagnosi precoce ed alla riabilitazione efficace del soggetto con un difetto uditivo. Il modello ideale prevede la creazione di centri multidisciplinari in cui si realizzi una perfetta integrazione tra specialisti di varie discipline e tra percorsi di approfondimento diagnostico di tipo strumentale e laboratoristico in continua evoluzione tecnologica. Questo dovrebbe portare in prima istanza ad un’accurata diagnosi audiologica corredata di tutta la documentazione relativa seguita dalla consulenza genetica e dai test molecolari. Un modello di integrazione tra audiologi/ORL e genetisti in un percorso clinico-diagnostico comune delle ipoacusie ereditarie sarà sempre più richiesto anche in considerazione del numero cerscente di screening neonatali universali di tipo audiologico eseguiti.
Le indagini genetiche cliniche, e strumentali, dovrebbero essere avviate solo dopo il completamento della diagnosi audiologica e dopo aver escluso le cause non genetiche. Al termine di questo primo percorso, il paziente dovrà essere indirizzato dal Clinico audiologo/ORL presso il centro di Genetica Medica con cui è strettamente in contatto, dove si procederà secondo la metodologia della consulenza genetica, che dovrà prevedere in prima istanza la raccolta di dati anamnestici, quindi l’esecuzione di un accurato esame obiettivo e la presa di visione dei risultati degli esami strumentali.
L’anamnesi familiare e personale mirerà ad evidenziare presenza di consanguineità nei genitori del probando, presenza di ipoacusie nei genitori o in collaterali, presenza di acufeni e gruppo etnico di appartenenza.
Queste informazioni porteranno a definire un modello probabile di trasmissione ereditaria della malattia.
Inoltre il Genetista dovrà prendere in considerazione i risultati delle indagini strumentali ivi incluse: caratteristiche audiometriche (forma neuro-sensoriale, trasmissiva, età di insorgenza, e funzione vestibolare). Infine verificherà che siano state escluse tutte le cause esogene di ipoacusia ovvero: storia di infezione intrauterina (TORCH), di meningite, ossigenazione extraxorporea con membrana (ECMO), storia di ipossia, esposizione prenatale all’alcool o a farmaci ototossici.
L’esame obiettivo dovrà prevedere la misurazione dei parametri auxologici (altezza, peso, circonferenza cranica), e la ricerca delle anomalie minori del volto che possono far sospettare (parte di) un quadro riconducibile ad una sindrome dismorfica. Dovrà essere condotta una valutazione descrittiva di tutto il corpo analizzando inizialmente la cute e gli annessi cutanei, la conformazione del cranio e del volto del paziente, l’impianto anteriore e posteriore dei capelli e le loro caratteristiche, nonchè valutando le varie regioni del volto in direzione cranio-caudale.
L’esame obiettivo ed il corrispondente corredo diagnostico strumentale dovrà, in particolare, consentire di individuare l’eventuale presenza di
- Anomalie oculari: eterocromia dell’iride, miopia, retinite pigmentosa, distacco di retina, cataratta precoce.
- Anomalie facio/cervicali: distopia dei canti interni, anomalie del padiglione auricolare, fossette pre-auricolari, cisti branchiali, palatoschisi, anomalie dentarie.
- Anomalie della cute: incanutimento precoce, ciocca di capelli bianchi sulla fronte, difetti di pigmentazione, pelle secca, cheratoderma.
Il genetista dovrà inoltre verificare, attraverso i dati diagnostico-strumentali in possesso od eventualmente ottenibili, la presenza di
- Anomalie endocrine: tireopatie, diabete.(vanno messi tra i disturbi funzionali)
- Anomalie cardiache: storia di svenimenti, sincopi e morti improvvise, aritmie, aumento dell’intervallo Q-T, cardiopatia congenita.
- Anomalie renali: malformazioni congenite, ematuria, proteinuria. Questi sono esami di funzione
- Anomalie scheletriche con particolare riguardo alla colonna cervicale
È infine importante ai fini di un corretto inquadramento diagnostico richiedere con la maggior precisione possibile l’epoca di raggiungimento delle principali tappe di sviluppo psicomotorio, ed eventualmente avere a disposizione una valutazione di livello psicointellettivo effettuata da un Medico specialista in Neuropsichiatria.
A questo punto si potranno verificare due condizioni:
a) identificazione di un soggetto con ipoacusia associata a difetti congeniti multipli e/o dismorfismi (sospetto quadro sindromico). Sarà in questo caso necessario realizzare le fotografie, previo consenso del soggetto e/o della famiglia, secondo lo schema della visita dismorfologica
b) identificazione di un soggetto con una perdita uditiva apparentemente isolata, cioè non associata ad altre manifestazioni cliniche (quadro non sindromico)
1) Pazienti con ipoacusia neurosensoriale senza evidenti segni sindromici che, alla luce dei risultati di un’adeguata valutazione clinico-strumentale, si dimostrano affetti da un quadro sindromico ben definito in cui la ipoacusia rappresenta un elemento diagnostico fondamentale della condizione (vedi alcuni esempi in tabella n.1). Per una parte di queste condizioni è oggi disponibile un esame genetico molecolare di conferma che può essere suggerito dopo l’inquadramento clinico.
Tabella 1:
Sindrome di Alport
Sindrome Branchio-Oto-Renale
Sindrome Jervell-Lange-Nielsen
Sindrome di Pendred
Sindrome di Usher
Sindrome di Waardenburg
Sindrome di Stickler
2) Pazienti con ipoacusia neurosensoriale non isolata in cui viene successivamente posta una diagnosi di sindrome malformativa sulla base di specifici elementi clinici e strumentali ed in cui la presenza di ipoacusia costituisce una potenziale anomalia associata al quadro sindromico stesso. L'ipoacusia in questo caso non costituisce un elemento diagnostico fondamentale (es. Sindrome di Noonan).
3) Pazienti con specifici quadri sindromici in cui la ipoacusia neurosensoriale rappresenta una potenziale complicanza associata che potrebbe essere identificata nell’ambito del percorso di follow-up assistenziale mirato alla singola condizione. In questo caso, quindi, la diagnosi di ipoacusia è successiva all’inquadramento diagnostico sindromico del bambino affetto (es. Sindrome di Cornelia de Lange).
Sul piano pratico, quindi si possono configurare, nell'ambito dell'attività del Centro di Genetica Medica, due possibili percorsi per il paziente affetto da perdita uditiva/sordità di tipo sindromico:
Soggetto con ipoacusia nell’ambito di un evidente quadro polimalformativo:
eseguirà per la prima volta una valutazione dismorfologica in un contesto ambulatoriale, per impostare adeguatamente l’iter più adatto all’inquadramento diagnostico generale, e successivamente, a seconda della diagnosi finale posta, verrà rivalutato per il monitoraggio delle possibili complicanze mediante Day-Hospital e/o ricoveri presso il Centro di Riferimento. Eseguirà i test genetici (citogenetici e/o molecolari) specifici per ciascuna patologia
Soggetto con sindrome malformativa e potenziale rischio di ipoacusia come complicanza nota del quadro sindromico:
verrà impostata una accurata valutazione della funzione uditiva e, qualora essa risultasse anomala, opportunamente indirizzato per trattamento
Forme non sindromiche
A questo punto esclusa una forma sindromica, al fine dell’ottenimento di una diagnosi eziologica corretta, a prescindere dal tipo di trasmissione genetica ipotizzata sulla base dell’albero genealogico quale recessiva, dominante o caso isolato, si dovrà :
-eseguire la analisi del gene GJB2 (intera sequenza delle zone trascritte e di splicing), mediante tecnica di sequenziamento diretto)
-se non c’è omozigosi o eterozigosi composta GJB2 (connessina 26) o se c’è eterozigosi GJB2, studiare la regione promotrice di GJB2 (incluso il primo esone non tradotto) e studiare per il gene GJB6 (connessina 30) la mutazione Delta (GJB6-D13S1830) al fine di escludere subito forme di doppia eterozigosi (digenia) .
La analisi della connessina 26 e 30 è indicata soprattutto (ma non esclusivamente) nelle forme ad insorgenza precoce, non progressive, gravi bilaterali, con audiogramma in caduta sulle frequenze acute.
Se l’analisi molecolare dei geni GJB2 e GJB6 risulta negativa è consigliabile l’esecuzione del test specifico per la mutazione mitocondriale A1555G (frequenza nella popolazione 1/1000). In particolare si raccomanda questo test quando:
La diagnosi di laboratorio per tale mutazione deve comunque tenere in considerazione che l’eteroplasmia potrebbe non essere identificata se al di sotto di una certa percentuale (30%), per problemi di carattere puramente tecnico, sia che si utilizzi la sequenza che la digestione enzimatica
In presenza di soggetti negativi per le mutazioni nel gene GJB2, dovranno inoltre essere eseguiti di volta in volta, tenendo conto sia del tipo di trasmissione ereditaria presente in famiglia che del grado di gravità della malattia stessa, esami specifici quali: indagini di funzionalità tiroidea, visita oculistica ed elettroretinogramma, ECG, ecografia renale, test neuroradiologici
Uno schema riassuntivo del processo diagnostico molecolare e del management del paziente è riportato qui di seguito
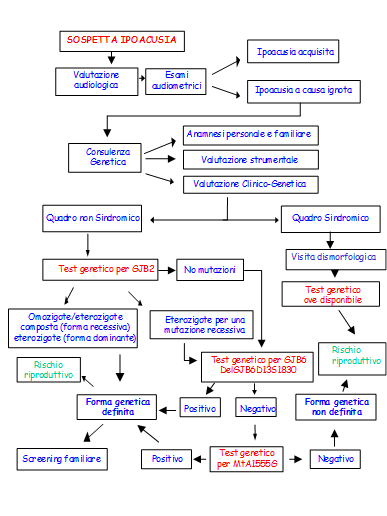
Se l’albero genealogico suggerisce una forma associata al cromosoma X per il momento ci si può limitare alla consulenza.
Altre specificità della consulenza e del management del paziente nelle forme isolate
a) La consulenza genetica che precede il test molecolare deve illustrarne le finalità, le sue limitazioni (dato l'elevato numero di geni associati a sordità), le sue caratteristiche e i possibili esiti. Trattandosi di un’indagine genetica è previsto che venga espresso un consenso scritto, distinto in caso di minori o di pazienti adulti.
b) Il referto di laboratorio deve essere chiaro ed universale (si intende l’utilizzo della nomenclatura ad esempio adottata nella pagina web della connessina 26 o nel sito The Hereditary Hearing Loss Homepage).
c) Nel caso di mutazioni nuove, sono necessarie le verifiche universalmente note nel campo della ricerca (verifica che non si tratti di polimorfismo, segregazione con la patologia, alterazione in zone conservate, ecc.), prima di attribuire alla alterazione molecolare un significato patologico.
d) In quei laboratori in cui si fa solo screening per alcune mutazioni più frequenti, è assolutamente necessario che si informi preventivamente, sia il paziente che il medico curante della limitatezza del test.
e) L’esito del test genetico, accompagnato da un referto scritto di consulenza genetica che precisi il rischio genetico di trasmissione sia in caso di test positivo che negativo e la sua limitatezza, deve essere consegnato dal genetista e spiegato al paziente (o ai genitori).
f) La diagnosi prenatale molecolare va resa disponibile alle coppie che dopo un accurata consulenza genetica e psicologica ne fanno richiesta.
g) I laboratori in cui si eseguono le diagnosi molecolari, dovrebbero essere certificati e/o accreditati presso le strutture regionali, per offrire un corretto iter, non solo diagnostico, ma anche di controllo del campione per quanto riguarda la sicurezza della privacy e la riduzione completa dell’errore diagnostico.
h) Sarebbe auspicabile, data la frequenza di questi esami genetici, si istituisse a livello nazionale un controllo di qualità specifico sul modello di quello della Fibrosi Cistica del Istituto Superiore di Sanità.
i) Sarebbe auspicabile un coordinamento tra i diversi laboratori di biologia molecolare, affinché si realizzasse una rete sul territorio nazionale per lo studio dei casi negativi ai test connessina e mitocondriale, con la selezione delle famiglie da sottoporre ad indagini per geni più rari. Il Coordinatore della presente Commissione, svolgendo già tale attività per alcuni centri italiani e diversi centri esteri, si dichiara disponibile ad estendere tale attività di coordinamento a tutti i centri nazionali.
Screening molecolari neonatali e/o del portatore
L’introduzione di tecnologie avanzate per la diagnosi molecolare delle perdite uditive quali ad esempio i microarray, apre la possibilità di pianificare screening molecolari per il gene GJB2. Difatti la disponibilità di test economici, accurati e standardizzati va a completare una situazione già di per se favorevole agli screening del gene GJB2 e caratterizzata da: a) presenza di una mutazione molto frequente nei caucasici, e di altre molto frequenti in altre popolazioni (mutazioni popolazione-specifiche), b) elevata frequenza dei portatori sani di queste mutazioni nella popolazione generale, b) elevata numerosità di soggetti affetti omozigoti per queste mutazioni. Stante la situazione possiamo ipotizzare due tipi di screening molecolare: 1) quello volto all’identificazione precoce dei soggetti affetti e 2) quello volto all’identificazione dei poratori sani nella popolazione generale.
Bibliografia di riferimento
Robertson ND, Morton CC, Beginning of a molecular era in hearing and deafness. Clin Genet 1999 55:149-159.
Denoyelle F, Marlin S, Weil D, Moatti L, Chauvin P, Garabedian E, Petit C. Clinical features of the prevalent form of childhood deafness, DFNB1, due to connexin-26 gene defect: implications for genetic counselling. Lancet 1999 353:1298-1303
Zelante L., Gasparini P., Esvill X. Connexin 26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Hum Mol Genet 1997, 6:1605-1609.
Gorlin R G. Hereditary hearing loss and its Syndromes. Oxford University Press, New York 1995.
Robn N.H., Smith R.J., Matthews A.L.Genetic Testing for Deafness in Clinical Practice. Audiological Medicine 2003; 1:89-93
King J I Ethical issues in the genetic study of deafness. Ann N Y Acad Sci 1991 630:236-239.
The Hereditary hearing Loss Homepage at http://dnalab-www.uia.ac.be/dnalab/hhh/
Dallapiccola B. Aspetti genetici delle sordità. Acta Otoringol Ital 1996, 16(2):79-90
Middleton A, Hewison J, Mueller R.F. Attitudes of deaf adults toward genetic testing for hereditary deafness. Am J Hum Genet 1998, 63: 1175-1180.
Brunger J.W.,Murray G.S.,O’Riordan M.A.,Mattews A.L. Smith RJH, Robin N.H. Parental attitudes towards genetic testing for pediatric deafness. American Journal of Human Genetics 2000; 67:1621-1625.
Del Castillo et al.: A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med. 2002 Jan 24;346(4):243-9
White K.R. Early Hearing Detection and Intervention Programs: Opportunities for Genetic Services. Am J Med Genet 130A:29-36 2004
Taneja P D Pandya A Attitudes of Deaf Individuals Towards Genetic Testing Am J Med Genet 130A: 17-21 2004
Fonte: http://www3.unisi.it/ricerca/dottorationweb/genetica_medica/News/Approved%20documents/Sordita-documento%2004.doc
Sito web da visitare: http://www3.unisi.it
Autore del testo: sopra indicato nel documento di origine
Il testo è di proprietà dei rispettivi autori che ringraziamo per l'opportunità che ci danno di far conoscere gratuitamente i loro testi per finalità illustrative e didattiche. Se siete gli autori del testo e siete interessati a richiedere la rimozione del testo o l'inserimento di altre informazioni inviateci un e-mail dopo le opportune verifiche soddisferemo la vostra richiesta nel più breve tempo possibile.
I riassunti , gli appunti i testi contenuti nel nostro sito sono messi a disposizione gratuitamente con finalità illustrative didattiche, scientifiche, a carattere sociale, civile e culturale a tutti i possibili interessati secondo il concetto del fair use e con l' obiettivo del rispetto della direttiva europea 2001/29/CE e dell' art. 70 della legge 633/1941 sul diritto d'autore
Le informazioni di medicina e salute contenute nel sito sono di natura generale ed a scopo puramente divulgativo e per questo motivo non possono sostituire in alcun caso il consiglio di un medico (ovvero un soggetto abilitato legalmente alla professione).
"Ciò che sappiamo è una goccia, ciò che ignoriamo un oceano!" Isaac Newton. Essendo impossibile tenere a mente l'enorme quantità di informazioni, l'importante è sapere dove ritrovare l'informazione quando questa serve. U. Eco
www.riassuntini.com dove ritrovare l'informazione quando questa serve