I riassunti , gli appunti i testi contenuti nel nostro sito sono messi a disposizione gratuitamente con finalità illustrative didattiche, scientifiche, a carattere sociale, civile e culturale a tutti i possibili interessati secondo il concetto del fair use e con l' obiettivo del rispetto della direttiva europea 2001/29/CE e dell' art. 70 della legge 633/1941 sul diritto d'autore
Le informazioni di medicina e salute contenute nel sito sono di natura generale ed a scopo puramente divulgativo e per questo motivo non possono sostituire in alcun caso il consiglio di un medico (ovvero un soggetto abilitato legalmente alla professione).
Farmacologia generale
Definizione di farmaco:
sostanza che, introdotta nell'organismo, è capace di modificarne la funzione. Non dà soltanto effetti benefici, ma anche negativi.es morfina: allevia il dolore ma dà anche sedazione, prurito, depressione respiratoria, morte.
Gli effetti benefici sono presenti solo in condizioni di patologia (es bradicardia da digitale in un soggetto normale è fugace, nel soggetto con insufficienza cardiaca la riduzione della frequenza diventa stabile).
Ci sono dosi che hanno effetti terapeutici e dosi con effetti indesiderati-> a determinate concentrazioni i farmaci sono terapeutici mentre a dosi maggiori possono essere veleni.
I farmaci possono essere->causali-> eliminano la causa (antibiotici)
-> patogenetici-> riducono gli effetti (antipertensivi)
-> sintomatici-> riducono o eliminano i sintomi (farmaci della terapia del dolore)
Effetto farmacologico e risposta al farmaco:
es la noradrenalina agisce sui recettori alfa (++) e beta (+). Se somministro la noradrenalina all'organismo in toto osservo un rallentamento della frequenza cardiaca perché la noradrenalina è molto più attiva sui recettori alfa e provoca vasocostrizione periferica con aumento della pressione diastolica e attivazione degli stimoli vagali con un rallentamento cardiaco(effetto farmacologico). Se io somministrasse la noradrenalina solo a livello cardiaco avrei un aumento della frequenza cardiaca(risposta al farmaco).???
placebo: in letteratura è definita come una sostanza priva di attività farmacologica specifica, somministrata per controllo nei test clinici, oppure ad un particolare paziente per stimolarne potenziali benefici psicologici.
Placebo background: rumori di fondo su cui si stabilisce l'effetto del farmaco
La medicina alternativa spesso si basa anche (o solo) sull'effetto placebo ma è stato dimostrato avere rilevanza nella terapia analgesica: il placebo può ridurre l'intensità del dolore anche del 35%. Il placebo si dimostra più evidente nei casi in cui la componente psicologica è preponderante-> dolore, ipertensione, depressione.invece ad esempio nel diabete l'effetto placebo è quasi inesistente.
L'azione analgesica da placebo è strettamente legata alla risposta fisiologica alle endorfine: persone che credevano di ricevere morfina ma invece ricevevano placebo rispondevano come se avessero preso morfina. Somministrato naloxone (blocca i recettori per il Oppioidi endogeni) l'effetto placebo scompariva-> conferma di relazione diretta tra endorfine e risposta al placebo
Condizionamento pavloviano
stimolo incondizionato stimolo condizionato
risposta incondizionata risposta condizionata
La risposta condizionata ha un effetto rapidissimo per l'apprendimento associativo. La fase di biodisponibilità è più rapida ma meno intensa nell'effetto, dal punto di vista clinico può essere sufficiente.
Farmaceutica + farmacocinetica + farmacodinamica
somministrazione del farmaco
Disgregazione del composto
solubilizzazione di principi attivi
= >I fase farmaceutica
Farmaco disponibile per l'assorbimento
Disponibilità farmaceutica
Assorbimento
distribuzione
metabolismo
escrezione/accumulo
= >II fase farmacocinetica (destino del farmaco nell'organismo)
Farmaco disponibile per l'azione
Disponibilità biologica
Azione sui recettori nei tessuti bersaglio
= >III fase farmacodinamica
Effetto
Farmacodinamica:
modalità attraverso cui il farmaco esprime i suoi effetti dal macro al micro all'interno dell’organismo.
Se Farmaco ha centro di chiralità->un isomero attivo, l'altro molto meno attivo
Es solo levonoradrenalina (idrossile in posizione sinistra) è in grado di interagire con il recettore-> forma attiva
Il farmaco induce il suo effetto interagendo con il proprio recettore:
tale legame può avere forza diversa:
Forza del legame farmaco-recettore
legame covalente (100 kcal/mole)
legame ionico 5 kcal/mole)
legame idrogeno (2-5 kcal/mole)
legame di Van der Waals (0,5 kcal/mole)
[La forza di legame può essere aumentata ad esempio sostituendo un idrossile con il fluoro (es Prozac)-> >affinità al sito di legame->stabilizzazione ]
Generalmente il legame è abbastanza debole->reversibile,ma in determinate situazioni nn lo è:
Legame covalente-irreversibile
Esteri organofosforici con colinesterasi (gas nervini)
Aspirina -piastrina (-> non più in grado di sintetizzare il trombossano)
[Legame ionico
I metalli (chelanti) sono in grado di formare complessi idrosolubili con varie sostanze favorendone l’eliminazione-> usati in ematologia e al pronto soccorso per inattivare sostanze tossiche.
EDTA-Ca impedisce al sangue di coagulare nella provetta
sottrae il piombo, rame, Mercurio, arsenico
Penicillamina-Rame nel morbo di Wilson
desferrossamina-Fe nell’emosiderosi
Altri metodi di detossificazione:
adsorbimento con carbone attivo: ogni particella ha una carica elettrostatica che attrae tutte le molecole portandole via-> utilizzato per la lavanda gastrica
trasportatori per i mediatori trans sinaptici
il mediatore rilasciato (es dopamina, noradrenalina ecc) nello spazio sinaptico viene ricaptato e riutilizzato
DAT: recupera dopamina e viene inibito dalla cocaina
trasportatore della serotonina, la sua attività è implicata nella patogenesi della depressione e dell'alcolismo. Inibito dal prozac e da altri antidepressivi.]
Tipi di Recettori
Recettori intracell per agenti liposolubili
Es No->guanililciclasi->GMPc
Corticosteroidi,h sessuali,vitamina D,ormoni tiroidei->Sequenze di dna specifiche->regolazione espressione geni vicini ecc
Recettori di membrana
Canali Ionici
-Recettori-canali (o recettori ionotropici)
rec nicotinico per Ach:
Canale pentamerico(ognuna delle cinque subunità è costituita da quattro catene (M1-M4)-leg cn Ach->modificaz conformazionale->apertura canale centrale->ingresso Na->depolarizzazione
rec NMDA(N-metil-D-aspartato) per Glutammato
costituito da due subunità (N1+N2)-su una vi sono siti di legame per glicina e serina, sull'altra per glutammato e aspartato (tutti attivatori). +sito di legame per il magnesio=>treno di stimoli (non uno solo)->magnesio salta-> apertura canale-> fluisce calcio all'interno della cellula-> modificazione responsabilità della zona che intorno recettore-> facilitazione trasmissione di sinaptica glutammatergica + fenomeno di Long Term Potentiation(facilitazione sinaptica a lungo termine)
-Canali voltaggio dipendenti es per Na,Ca, K
Canali del calcio voltaggio dipendenti:
tipoL: in tessuto muscolare liscio (es vasi), lunga durata d'apertura, elevata conduttanza, alta soglia di attivazione-> bloccato da Nifedipina
tipo T: in cuore e neuroni, breve durata d'apertura, basta conduttanza, bassa soglia di attivazione->bloccato da Flunarizina
tipo N: in neuroni, basta durata d'apertura, alta soglia di attivazione->bloccato da Conotossina
tipo P: in neuroni, cellule di Purkinje, lunga durata d'apertura, alta soglia di attivazione->bloccato da Conotossina
Canali del sodio voltaggio dipendenti:
in assone neuronale,cardiomiociti->cambiamento voltaggio->apertura canale->afflusso Na->depolarizzazione-> potenziale d’azione
presente in diversi stati (chiuso, aperto, inattivato-> bersaglio non sempre reattivo nei confronti dei farmaci)
può avere sito di legame della lidocaina che si lega a fenilalanina e tirosina che costituiscono il canale stesso
Recettori metabotropici:
Effetti adattativi a esposizioni ai farmaci:
desensibilizzazione:non tutti uguali.un meccanismo è quello dei rec Beta adrenergici:
legame con rec->modificaz conformazionale->affinità a protein chinasi beta ark->fosforilazione residui treoninici e serinici->>affinità a legame con beta arrestina-><affinità a proteina G.termina appena non c’è + agonista a stimolare
down regulation recettori:internalizzazione farmaco recettori->lisosomi->degradazione recettori-><numero su membrana. Termina dopo un certo periodo di tempo che non c’è + agonista
La relazione dose-risposta in vivo è piuttosto complessa.
Invece nei sistemi in vitro la relazione tra concentrazione del farmaco e l'effetto è descritta da una curva iperbolica con equazione
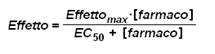
Che può essere graficata anche in forma semilogaritmica in modo da espandere la scala alle basse concentrazioni(dove l’effetto varia velocemente) e comprimerlo alle alte(dove varia lentamente)->vedo l’effetto a diversi ordini di grandezza.
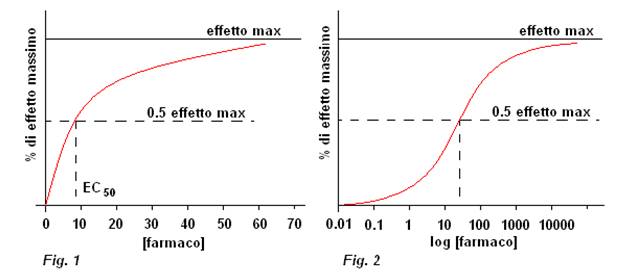
Questa relazione iperbolica somiglia a quella della legge di azione di massa,ovviamente perché il legame farmaco rec responsabile è caratterizzato dalla stessa cinetica di saturazione.
->Un grafico che descrive la percentuale di legame al recettore in funzione dell’aumentare del ligando è sovrapponibile al precedente
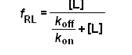
->Koff/Kon=Kdissociazione=conc farmaco in cui 50%ligando legato e 50%libero=indica affinità di un farmaco per un determinato recettore(alta Kd->bassa affinità)
EC50=Kd se agonista pieno e no recettori di riserva
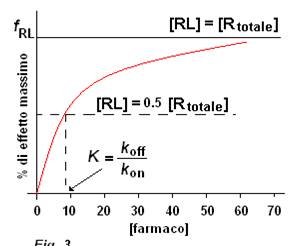
Potenza di un farmaco: dipende dalla concentrazione o dose del farmaco necessaria a produrre una risposta uguale al 50% della risposta massima->se EC50 alta->potenza bassa e viceversa
Efficacia: parametro che riflette il limite della relazione dose-risposta(effetto massimo); è determinata dal modo di interagire del farmaco con recettore o dalle caratteristiche del sistema recettore-effettore in gioco.
[I farmaci sono selettivi piuttosto che specifici nelle loro azioni poiché legano uno o più recettori più selettivamente rispetto ad altri con induzione di effetti ben distinti. La selettività di un farmaco si valuta separando i suoi effetti in effetti terapeutici e tossici
effetti terapeutici e tossici dati dallo stesso meccanismo recettore-effettore (insulina-> coma ipoglicemico)
effetti terapeutici e tossici mediati da = recettori posti su tessuti diversi (glicosidi digitalici-> inibizione Na/K ATPasi)
effetti terapeutici e tossici mediati da recettori di tipo diverso.]
Effetto dell’interazione farmaco-recettore
dipende dall'affinità del farmaco per il recettore
dall'attività intrinseca del farmaco
i farmaci sono dotati di bassa affinità e di attività intrinseca variabile
distinzione dei farmaci in base all'effetto ottenuto nel legame con il recettore
agonista pieno-> il legame modifica la funzione cellulare
agonista parziale-> minore attività intrinseca rispetto all'agonista pieno -> effetti opposto visibile soltanto in presenza dell'agonista pieno.
antagonista-> il legame non altera la funzione cellulare-> gli effetti sono visibili soltanto in presenza di agonisti che sono impossibilitati a svolgere la loro funzione
agonista inverso parziale-> induce effetti contrari a quelli dell'agonista parziale
agonista inverso pieno -> induce effetti contrari a quelli dell'agonista pieno (es benzodiazepine:effetto ansiolitico legandosi a rec GABA;si pensa ci sia 1 agonista inverso endogeno che causa ansia)
Antagonismo competitivo:antagonista competitivo si lega reversibilmente allo stesso recettore dell’agonista e ne diminuisce l’affinità->aumentando concentrazione di agonista è possibile ottenere cmq effetto massimale(curva dose risposta logaritmica si sposta a dx ma nn viene deformata,curva dose risposta lineare viene deformata,ma l’altezza è =)
NB il gioco tra agonisti e antagonisti fa si che si possano somministrare concentrazioni dell’uno e dell’altro che da soli risulterebbero letali
Il rapporto tra la concentrazione di agonista necessaria a dare un certo effetto in presenza d antagonista e la concentrazione necessaria a dare lo stesso effetto in assenza di antagonista (=rapporto di dose) dipende dalla concentrazione di antagonista e dalla sua Kd mediante l’equazione di Schild:
Rd=1+ [I]/Ki
Antagonismo non competitivo:antagonista non competitivo si lega irreversibilimente allo stesso recettore-> non si può + raggiungere effetto massimale anche aumentando le concentrazioni dell’agonista->curva dose-risposta viene schiacciata verso il basso ma non verso dx->EC50 rimane la stessa
[Altri tipi di antagonismo:chimico->farmaco antagonizza l’altro direttamente senza intervenire su rec;fisiologico->az d un farmaco antagonizza azione dell’altro)
Ipotesi dei recettori risparmiati (o di riserva)
si parla di recettori si di riserva quando per un certo effetto farmacologico il massimo della risposta può essere ottenuto con concentrazioni di agonista insufficiente ad occupare tutti i recettori disponibili-> anche se è presente un antagonista non competitivo si può ottenere comunque un effetto massimale fino ad un certo punto.
Questo probabilmente è dovuto ad un'ampia efficienza dell'accoppiamento recettore-effettore.
Un volta che l’antagonista non competitivo avrà occupato la riserva recettoriale l’agonista non sarà + in grado di indurre un effetto massimale anche aumentandone le concentrazioni.
[es acetilcolina-> agonista
atropina-> antagonista competitivo dell'acetilcolina
se presenti entrambi si può ottenere l'effetto massimale fino a che l’ atropina non aumenta tanto da intaccare la riserva dei recettori.
es di applicazione clinica: intossicazione da esteri fosforici (gas nervini, anticrittogamici) bloccano in maniera irreversibile l'acetilcolina esterasi e passano la barriera e ematoencefalica-> usando dosi massive di atropina è possibile impedire il legame del recettore con l'acetilcolina-> si antagonizza l'effetto degli esteri fosforici ]
Curve dose-effetto quantali:
le curve graduali non sono utili se il mio effetto è del tipo tutto o nulla(es morte) o se in campo clinico l’effetto varia da persona a persona e voglio vedere come->
curve quantali
Indice terapeutico: rapporto tra dose di farmaco richiesta per produrre un effetto desiderato nel 50% della popolazione e dose di farmaco che produce un effetto indesiderato nel 50% della popolazione
Farmacocinetica
Meccanismi di attraversamento delle membrane biologiche
farmaci liposolubili ->diffusione passiva (principio di Meyer Hoverton per gli anestetici generali: più sono lipofili più passano la barriera)
elettroliti deboli (maggioranza dei farmaci)-> forma indissociata per diffusione passiva
farmaci idrofili non ionizzabili di piccole dimensioni (< 4 a)-> filtrazione attraverso pori
Farmaci idrosolubili non ionizzabili con diametro maggiore di 4 a-> diffusione facilitata senza dispendio di energia mediante trasportatore
acidi e basi organiche ionizzate-> trasporto attivo con dispendio energetico mediante un trasportatore
proteine e altre grosse molecole-> fagocitosi e pinocitosi (trasporto vescicolare)
Effetti del Ph sull'attraversamento delle membrane biologiche
elettrolita debole attraversa la barriera in forma indissociata. Il trasferimento dipende da pka della sostanza e dal gradiente di Ph ai due lati della membrana.
Legge di azione di massa
Es aspirina ha pKa uguale 4,4-> se la somministro a stomaco vuoto-> forma indissociata-> passa facilmente la mucosa dove trova Ph = 7,4-> si comporta come forte donatore di protoni-> prevale la forma dissociata in circolo. Questa non passa la barriera ematoencefalica e viene filtrata dal rene. Se c'è condizione di acidosi il Ph scende-> aumento quota indissociata-> superamento barriera ematoencefalica (-> inibizione dei centri respiratori) e riassorbimento renale-> circolo vizioso.per romperlo: diuresi forzata e alcalinizzazione.
Per i farmaci basici viceversa.
Parametri farmacocinetici:
*volume di distribuzione
indica la capacità di diffusione e penetrazione del farmaco nei vari organi tessuti dell'organismo.
= dose somministrata/concentrazione di farmaco nel sangue
Distribuzione possibile:
solo plasma
fluidi extracellulari
fluidi intracellulari(dove può anche concentrarsi aumentando ulteriormente il volume di distribuzione)
*Biodisponibilità:
entità dell'esposizione sistemica ottenuta dopo somministrazione per una qualsiasi via di somministrazione rispetto alla somministrazione endovenosa di una stessa dose di farmaco
anche se teoricamente la via endovenosa è al 100% disponibile potrebbe non essere così per
legame farmaco proteico:le proteine plasmatiche hanno gruppi funzionali che legano sostanze e endogene ma che possono legare anche farmaci->< biodisponibilità es fans,warfarin
I fattori che modificano il legame farmaco proteico: condizioni che alterano il tasso di proteine plasmatiche (insufficienza epatica, insufficienza renale, enteropatie, parassitosi, ustioni)
NB se albumina si riduce molto-> farmaco non legato aumenta molto MA anche ascite in cui il farmaco si può accumulare
*Clearance plasmatica
volume di plasma depurato dal farmaco nell'unità di tempo.
Cltot =Cl renale +Cl non renale (perlopiù epatica)
quando la clearance totale > 650 ml al minuto(=Cl renale max)-> alla sua eliminazione concorre in modo significativo il fegato
*Emivita plasmatica di eliminazione (T1/2)
tempo necessario affinché la concentrazione plasmatica si riduca del 50%
= 0,693 x. volume di distribuzione/clearance
tempo necessario alla completa eliminazione dall'organismo: 4-5 volte l’ emivita
Farmaci idrosolubili i: t/2 aumenta se versamento pleurico o insufficienza renale
farmaci liposolubili:t/2 aumenta se obesità o insufficienza epatica
*Concentrazione plasmatica di stato stazionario(=steady state)
durante una terapia di mantenimento il farmaco tende ad accumularsi nell'organismo ed i suoi livelli aumentano gradatamente fino a raggiungere uno stato di equilibrio o stato stazionario quando la quantità di farmaco eliminata nel tempo che intercorre tra una somministrazione dell'altra è uguale alla dose (avviene dopo circa 4-5 volte il t/2 del farmaco).
Es se un farmaco ha un’emivita di 12 h,lo somministrerò ogni 12 h, dopo quattro somministrazioni arriverò allo stato stazionario.
se voglio raggiungere più rapidamente concentrazioni terapeutiche simili a quelle dello stato stazionario devo fare
dose da carico= volume di distribuzione x concentrazione plasmatica desiderata.
Questo vale per i farmaci a somm orale perché quelli ev arrivano allo steday state più rapidamente->nn necessaria la dose da carico.
*Indice terapeutico:
dose letale nel 50% degli animali trattati/dose efficace nel 50% dei casi
o
dose tossica nel 50% sogg trattati/dose efficace nel 50% dei casi
l'indice terapeutico sarà tanto più basso quanto più il farmaco è potenzialmente tossico es digitale
Fasi della farmacocinetica di un farmaco:
Assorbimento:
passaggio del farmaco dalla sede di somministrazione al torrente circolatorio sistemico
Vie di somministrazione:
*via sublinguale
passaggio diretto ( evitando il filtro epatico) nella circolazione sistemica
pro:
azione terapeutica si manifesta più rapidamente che per via orale
no effetto di primo passaggio
particolarmente utile per nitroglicerina nelle crisi anginose perché il circolo buccale è tributario della vena cava superiore-> arriva direttamente al cuore
possibilità di sputare la compressa quando non più necessaria
possibilità di rapida automedicazione
MA incertezza di dosaggio
Indicazioni:analgesici,nitroderivati
[il filtro epatico può essere un vantaggio se voglio proteggere la via sistemica:spray cortisonico in crisi asmatiche: un po' va nelle vie respiratorie dove sortisce il suo effetto e un po' nel digerente. Viene poco assorbito ma ciò che passa al sangue ha un alto effetto di primo passaggio-> via sistemica protetta]
*Somministrazione transcutanea
applicazione di cerotto con serbatoio di farmaco-> il farmaco passa lo strato corneo-> derma dove si accumula e lo satura, tanto farmaco passa al derma tanto va in circolo
nell'arco di tempo la somministrazione è costante
indicazioni: terapie ormonali, nitroderivati, cloridrina (ipertensione)
*Via orale
sede di assorbimento influenzata da Ph:
acidi deboli-> assorbimento gastrico
quasi deboli-> assorbimento intestino tenue
Biodisponibilità influenzata da
-natura del farmaco (proteici o mucopolisaccaridici degradati prima di assorbimento, idrofili o substrati di proteina P e/o cyp450 non assorbiti)
-formulazione del farmaco: da max a min- soluzione, sospensione, capsula, compressa, compressa rivestita
soluzione-> dipende da tempo di svuotamento gastrico perché assorbimento perlopiù a livello del tenue
sospensione acquosa (in farmaco insolubili in acqua)-> velocità di dissoluzione
capsule e compresse:-> tempo di disintegrazione e dissoluzione, grado di compressione e dimensione delle particelle, tempo di svuotamento gastrico per capsule gastroresistenti
-presenza o meno di cibo nel lume:
es digitale +>> fibre
tetracicline + calcio, ferro, alluminio, magnesio
farmaci lipofili + alimenti->> biodisponibilità
-alterazioni della motilità intestinale
-effetto di primo passaggio (perlopiù epatico a livello microsomiale, ma anche intestinale-flora batterica, succo gastrico, enzimi della parete intestinale, glicoproteina P, citocromo P 450)
buona compliance
ma lento assorbimento-> no in situazioni di emergenza
*Via rettale:
biodisponibilità influenzata da
formulazione del farmaco (da max a min: liquida , supposta lipofila, supposta idrofila)
mucosa non ben assorbente
plesso emorroidario: tributario della vena cava inferiore e della vena porta
Ok per farmaci sintomatici e in pazienti pediatrici o con alterazioni funzionali gastroenteriche
*Via inalatoria
assorbimento molto rapido
ok per anestesia generale, situazioni di emergenza
*Via endovenosa
per bolo
per infusione intermittente (alcune decine di minuti)
continua (24 h su 24)
effetto rapido
possibilità di diluire i farmaci
*Via intramuscolo e sottocute
biodisponibilità e velocità di assorbimento influenzate da
Ph, volume, osmolarità, viscosità
maggiore rapidità di assorbimento rispetto alla via orale
possibilità di somministrare farmaci diluiti in piccoli volumi
Distribuzione
passaggio del farmaco dal torrente circolatorio ai diversi organi e tessuti.
Il circolo può essere anche il punto di azione del farmaco:
aspirina come antiaggregante
nitroderivati (endotelio, muscolatura liscia)
coxib: non inibizione cox 1-> non gastrolesivi
inibizione cox 2 (costitutive dell'endotelio basale, sintesi prostraci cline = vasodilatatrici e antiaggreganti)-> protrombotici
Più comunemente il farmaci hanno azione al di fuori del torrente circolatorio.
Il processo distributivo dipende da
idro/liposolubilità
peso molecolare
legame farmaco proteico
caratteristiche anatomiche e vascolarizzazione dei vari distretti
Idrosolubile-> passaggio solo in spazi interstiziali
liposolubili-> diffusione passiva all'interno delle cellule
< peso molecolare, < legame farmaco proteico-> rapida distribuzione
> vascolarizzazione di un organo o tessuto-> rapida distribuzione
Barriere naturali: es ematoencefalica
vasi cerebrali con giunzioni serrate molto strette + glia che avvolge con podociti il vaso-> ostacolo al passaggio dei farmaci .
passaggio facilitato da lipofilia (pka lo influenza), < peso molecolare, presenza di gruppi particolari es sulfidrilico (es tiopentale) rispetto a ossidrilico (pentobarbitale): nonostante Ka sia maggiore nel pentobarbitale 1 min dopo la somministrazione di tiopentale C'è quantità maggiore in cervello che in plasma.tuttavia la concentrazione diminuisce rapidamente per retro diffusione nel plasma mentre quella del pentobarbitale aumenta più tardi ma declina molto lentamente.
alcuni farmaci non passano la barriera ematoencefalica perché pompati fuori dalla glicoproteina P (anche in cellule tumorali, microorganismi-> responsabile di resistenza al farmaco; in epatociti-> estrusione vile dal polo biliare; in tubulo renale-> estrusione attiva farmaci; in barriera emato testicolare; in linfociti-> importante in terapie antiretrovirali). La glicoproteina P ha una ricca variabilità di polimorfismo puntiforme.
Il compartimento di deposito di un farmaco può essere completamente diverso da quello di azione e dipende dal suo legame con costituenti come proteine, fosfolipidi, nucleo proteine (tropismo del farmaco per quel tessuto):
Tetracicline-> tessuto osseo(essendo fluorescenti provocano discromie al livello del tessuto dentale)
tiopentale (anestetico)-> tessuto adiposo
clorochina (antimalarico)-> fegato
amiodarone (antiaritmico)-> tiroide
Metabolismo
processi di biotrasformazione perlopiù enzimatica atta a trasformare farmaci liposolubili in metaboliti più idrosolubili che possano essere eliminati per via renale. Questi processi avvengono contemporaneamente all’ assorbimento e all'escrezione.
Avvengono tramite reazioni chimiche di:
ossidazione
riduzione
idrolisi ecc
= > fase 1 = funzionalizzazione-> sistema microsomiale,con cyp 450,svela un gruppo particolare della molecola-> la rende più idrosolubile, spesso inattiva.se la molecola non è abbastanza idrosolubile si passa alla fase 2.
glucuronazione
solfatazione
acetilazione
= > fase 2 = coniugazione-> trasferasi microsomiali o citosoliche aggiungono un substrato che trae origine dagli alimenti-> La coniugazione può dipendere anche molto dalla dieta.si ottengono molecole polari-> facilmente escrete.generalmente sono inattive ma a volte tossiche e, essendo idrosolubili, anche più nocive.
i metaboliti cosi ottenuti possono essere attivi o inattivi, tossici o meno.
es paracetamolo-> chinolone altamente reattivo-> se non neutralizzato dal glutatione-> processi ossidativi
Il centro del sistema di trasformazione è il citocromo P450: sistema ossido riduttivo da ione ferrico a ferroso con legame e metabolismo del farmaco lipofilo.
Diversi isoenzimi:
3A4 e 2d6-> affini a molti farmaci
2E1-> alcol e paracetamolo
Cyp450 può essere indotto o inibito:
inibitori (metaboliti ottenuti inattivano l'enzima)
amiodarone
imidazolici
cimetidina
macrolidi
isoniazide
fluorochinoloni
IPP
chinidina
succo di pompelmo (contiene derivati cumarinici e bioflavonoidi che danno inibizione intestinale irreversibile del cyp3a4 fino a ventiquattr'ore dall'assunzione->> assorbimento intestinale di molti farmaci)
Induttori (agiscono sull'espressione dei geni che servono alla sintesi del citocromo o ne inibiscono la degradazione)
carbamazepina
Fenobarbital (es con idrossicumarina = anticoagulante-> il tempo di protrombina cambia nonostante la dose sia costante)
fenitoina
Rifampicina
Grande polimorfismo per citocromo p450-> variabile velocità di metabolismo-> variabili concentrazioni plasmatiche del farmaco->poor e extensive metabolizer che necessitano di dosi di farmaco diverse
Eliminazione
* Via renale (la più frequente)
quota libera-> filtrazione glomerulare
se liposolubili-> riassorbimento per diffusione passiva
se idrosolubile-> no riassorbimento
se elettrolita debole-> forma dissociata non riassorbibile, forma indissociata riassorbibile-> alcalinizzo urine per eliminare il farmaci acidi e viceversa
NB la clearance renale è determinante per valutare le emivita del farmaco
*Via biliare
liposolubili-> diffusione passiva
idrosolubili-> trasporto attivo
*Via polmonare
gassosi (anestetici generali)e volatili (alcol etilico) -> diffusione passiva dal plasma all'aria alveolare
NB il farmaci liposolubili possono diffondere passivamente nel latte. Controindicazione all'allattamento al seno solo nel caso in cui farmaco in questione sia caratterizzato da un profilo di considerevole tossicità per il lattante.
Risposta al farmaco dipende da:
fattori inerenti al farmaco:
-formulazione farmaceutica
-schema terapeutico e compliance allo schema
Fattori inerenti al paziente
-Tolleranza
riduzione dell'effetto di una data dose di farmaco quando venga somministrata ripetutamente. Si sviluppa nei confronti degli effetti dei farmaci ottenuti a determinate dosi e non nei confronti del farmaco.La farmaco tolleranza è reversibile in un periodo di tempo più o meno breve;per ottenere effetto iniziale devo aumentare la dose.
se iposensibilità a effetto terapeutico: riduzione dell'indice terapeutico per necessità di aumentare la dose (es. morfina può essere sia analgesica che iperalgica.se tolleranza all'effetto analgesico prevale l'effetto iperalgico-> iperalgesia, allodinia (stimolo tattile che diventa doloroso)
se iposensibilità a effetto collaterale: aumento dell'indice terapeutico
Farmaco abitudine semplice: (antineoplastici, beta-bloccanti eccetera)-> scomparsa effetti positivi ma non disturbo, no desiderio del farmaco
farmacoabitudine voluttuaria (caffè, fumo)-> desiderio più o meno intenso, irritabilità, cattivo umore
abitudine tossicomanigena(alcol, Oppioidi, allucinogeni, psicostimolanti)-> sindrome di astinenza
Tolleranza acuta (Tachifilassi)-> tolleranza che si manifesta dopo poche dosi es cocaina
Tolleranza cronica (Bradifilassi)-> tolleranza che si manifesta dopo diverse dosi es lassativi, ipnotici
Tolleranza congenita
tolleranza acquisita di tipo farmacocinetico: cambiamenti nella distribuzione o nel metabolismo
tolleranza acquisita di tipo farmaco dinamico: riduzione della densità recettoriale, cambiamenti qualitativi dell'assetto recettoriale, perdita dell'affinità per l'agonista, incapacità di trasporre il segnale = > riduzione dell'effetto stimolante
Può essere causato da alterazioni metaboliche e funzionali (al livello del recettore = down regulation, della cellula e del sistema neurale che media la risposta )
-sensibilizzazione:
somministrando per un certo tempo antagonisti-> fenomeno dell’up regulation: > espressione recettori corrispondenti-> sospendendo i antagonisti->over shoot: risposta esagerata a concentrazioni fisiologiche di agonista. L'agonista può essere dannoso per ragioni opposte.
-fattori genetici
-fattori dietetici
-peso corporeo o, meglio, superficie corporea
> superficie-> > dose necessaria
NB in pazienti obesi i farmaci lipofili tendono ad accumularsi nel tessuto adiposo-> ritardo nella comparsa della risposta terapeutica, prolungamento dell'azione dopo l'interruzione del trattamento
-età
bambino piccolo: vie di eliminazione a capacità limitata
peristalsi intestinale irregolare, svuotamento gastrico rallentato, Ph gastrico più elevato
> sensibilità a farmaci e suscettibilità a reazioni avverse
anziani: < massa magra, < albumina, < acqua corporea, maggiore di tessuto adiposo-> alterata distribuzione dei farmaci
< funzionalità renale, < attività alcuni enzimi epatici-> clearance ridotta
-Sesso
differenze nell'attività di alcuni enzimi
diverso rapporto tra ormoni sessuali
differenza di peso corporeo
differenza di quantità di tessuto adiposo
periodo mestruale, menopausa, trattamento con contraccettivi orali, gravidanza
-stati patologici
insufficienza epatica
insufficienza renale
patologie gastrointestinali
insufficienza circolatoria: alterazioni di svuotamento gastrico, < motilità intestinale, < vascolarizzazione diversi distretti-> somministrazione orale poco indicata; meglio e endovenosa o endotracheale
Interazioni tra farmaci
interazioni farmaceutiche
es chinoloni + antiacidi-> somministrazione almeno a 6 h di distanza
L.-dopa + integratori di ferro
tetracicline + calcio, magnesio, alluminio, ferro
Interazioni farmacocinetiche
in fase di assorbimento
antiacidi + farmaci acidi (salicilati, alcuni antibiotici)->< assorbimento
antiacidi + farmaci basici (es antidiabetici orali)->> assorbimento
procinetici + diversi farmaci (es paracetamolo, Diazepam, alcol)->> velocità assorbimento
farmaci che riducono lo svuotamento gastrico + L-dopa, penicilline, digossina->< assorbimento
In fase di distribuzione
spiazzamento del legame farmaco proteico ad opera di altri farmaci con affinità maggiore. Effetto clinicamente rilevante se il farmaco spiazzato al legame farmaco proteico > 905% e a un basso indice terapeutico
es. Fans spiazzano anticoagulanti orali-> emorragia
antidiabetici orali-> episodico glicemici
In fase di biotrasformazione
induzione o inibizione dei citocromi
In fase di escrezione
competizione a livello della secrezione tubulare attiva
es Fans + metotressato
Risposte abnormi alla somministrazione dei farmaci
Reazioni idiosincrasiche
possono manifestarsi dalla prima somministrazione perché causate da un'alterazione genetica, sono imprevedibili e a volte molto gravi
dose dipendente, terapia da effettuarsi con antagonisti
con comparsa di effetti tossici da ridotta sintesi di enzimi, da sintesi di enzimi alterati, da proteine trasportatrici alterate
con mancata comparsa dell'effetto farmacologico atteso da alterazione della sintesi enzimatica, da alterazione dei recettori, da alterazioni di assorbimento
Reazioni allergiche:
si manifestano dopo la prima somministrazione (necessaria sensibilizzazione con il farmaco stesso o con farmaco simile nella struttura), sono imprevedibili, dose indipendenti, terapia con adrenalina, cortisonici, antistaminici
-Reazioni di tipo I (anafilattiche)
interazione IgE con superficie cellule presentanti l'antigene-> liberazione mediatori-> valuti dilatazione, edema, infiammazione-> orticaria, rinite, a mano, shock anafilattico-> mantenimento pervietà vie aeree, massaggio cardiaco, adrenalina, antistaminici a uno, cortisone
-Reazioni di tipo II (citolitiche)
interazione IgG e IgM con determinanti antigienici delle membrane cellulari-> attivazione complemento-> lisi cellule (emazie-anemia emolitica; neutrofili-granulocitopenia, piastrine-porpora trombocitopenica, cellule tubuli renali-nefropatie interstiziali acute
-Reazioni di tipo III (IC mediate)
immuno complessi IgG-antigene-> complemento-> deposito nell'endotelio vascolare-> risposta infiammatoria lesiva = malattia da siero uno a due settimane dopo l'inizio del trattamento: malessere, prurito, linfoadenopatia, artralgie, rinite, congiuntivite, vomito, diarrea
polmoniti da ipersensibilità
vasculite
-Reazioni di tipo IV (cellulomediate o ritardate)
linfociti T. e macrofagi + antigene-> produzione linfochine-> richiamo neutrofili ematofagi-> dermatite da contatto, febbre da farmaci, stomatite allergica
Fonte: http://www.accentosullad.com/public/simple/index.php?action=dlattach;topic=1514.0;attach=833
Sito web da visitare: http://www.accentosullad.com/
Autore del testo: non indicato nel documento di origine
Il testo è di proprietà dei rispettivi autori che ringraziamo per l'opportunità che ci danno di far conoscere gratuitamente i loro testi per finalità illustrative e didattiche. Se siete gli autori del testo e siete interessati a richiedere la rimozione del testo o l'inserimento di altre informazioni inviateci un e-mail dopo le opportune verifiche soddisferemo la vostra richiesta nel più breve tempo possibile.
I riassunti , gli appunti i testi contenuti nel nostro sito sono messi a disposizione gratuitamente con finalità illustrative didattiche, scientifiche, a carattere sociale, civile e culturale a tutti i possibili interessati secondo il concetto del fair use e con l' obiettivo del rispetto della direttiva europea 2001/29/CE e dell' art. 70 della legge 633/1941 sul diritto d'autore
Le informazioni di medicina e salute contenute nel sito sono di natura generale ed a scopo puramente divulgativo e per questo motivo non possono sostituire in alcun caso il consiglio di un medico (ovvero un soggetto abilitato legalmente alla professione).
"Ciò che sappiamo è una goccia, ciò che ignoriamo un oceano!" Isaac Newton. Essendo impossibile tenere a mente l'enorme quantità di informazioni, l'importante è sapere dove ritrovare l'informazione quando questa serve. U. Eco
www.riassuntini.com dove ritrovare l'informazione quando questa serve